

2.2. Химический состав и формулы минералов
2.3. Физические свойства минералов
Морфологические особенности кристаллов минералов
Блеск и показатель преломления
Когда мы осматриваем минералы в музейных витринах или лотках со специально подобранными образцами, нас невольно поражает то разнообразие внешних признаков, по которым они отличаются друг от друга.
Одни минералы кажутся прозрачными (горный хрусталь, каменная соль), другие — мутными, полупрозрачными или совершенно не пропускающими свет (магнетит, графит).
Замечательной особенностью многих природных соединений является их окраска. Для ряда минералов она постоянна и весьма характерна. Например, киноварь (сернистая ртуть) всегда обладает карминно-красным цветом, для малахита характерна ярко-зеленая окраска, кубические кристаллики пирита легко узнаются по металлически-золотистому цвету и т. д. Наряду с этим окраска большого количества минералов изменчива. Таковы, например, разновидности кварца: бесцветные (прозрачные), молочно-белые, желтовато-бурые, почти черные, фиолетовые, розовые.
Блеск — также весьма характерный признак многих минералов. В одних случаях он очень похож на блеск металлов (галенит, пирит, арсенопирит), в других — на блеск стекла (кварц), перламутра (мусковит). Немало и таких минералов, которые даже в свежем изломе выглядят матовыми, т. е. не имеют блеска.
Часто минералы встречаются в кристаллах, иногда очень крупных, иногда чрезвычайно мелких, устанавливаемых лишь с помощью лупы или микроскопа. Для ряда минералов кристаллические формы весьма типичны, например, для пирита — кубические кристаллы, для гранатов — ромбические додекаэдры, для берилла — шестигранные призмы. Однако в большинстве случаев минеральные массы наблюдаются в виде сплошных зернистых агрегатов, в которых отдельные зерна не обладают кристаллографическими очертаниями. Многие минеральные вещества распространены также в виде «натечных» масс, иногда причудливой формы, ничего общего не имеющей с кристаллами. Таковы, например, почковидные массы малахита, сталактитоподобные образования лимонита (гидроокислов железа).
Минералы различаются и по другим физическим свойствам. Одни из них настолько тверды, что легко оставляют царапины на стекле (кварц, гранат, пирит); другие сами царапаются обломками стекла или острием ножа (кальцит, малахит); третьи обладают настолько низкой твердостью, что легко чертятся ногтем (гипс, графит). Одни минералы при раскалывании легко расщепляются по определенным плоскостям, образуя обломки правильной формы, похожие на кристаллы (каменная соль, галенит, кальцит); другие дают в изломе кривые, «раковистые» поверхности (кварц). Широко варьируют и такие свойства, как удельный вес, плавкость и др.
Столь же различны и химические свойства минералов. Одни легко растворяются в воде (каменная соль), другие растворимы лишь в кислотах (кальцит), третьи устойчивы даже по отношению к крепким кислотам (кварц). Большинство минералов хорошо сохраняются в воздушной среде. Однако известен ряд природных соединений, легко подвергающихся окислению или разложению при действии кислорода, углекислоты и влаги, содержащихся в воздухе. Давно установлено также, что некоторые минералы под воздействием света постепенно меняют свою окраску.
Все эти свойства минералов, как мы теперь все больше и больше убеждаемся, находятся в причинной зависимости от особенностей химического состава и кристаллической структуры вещества — от конституции минерала, что в свою очередь обусловлено размерами атомов или ионов (участвующих в составе минерала), строением их электронных оболочек (особенно наружных) и свойствами, которые определяются положением химических элементов в системе Д. И. Менделеева. Поэтому многое из того, что раньше казалось загадочным, теперь, в свете современных достижений точных наук, становится все более и более понятным. Эти достижения способствуют не только правильному пониманию природных явлений, но и помогают нам в практическом использовании свойств минералов.
Конституция минерала представляет собой единство его химического состава и кристаллической структуры. Понятие «конституция» описывает, можно сказать, сущность минерала; она является его собственным, внутреннейшим свойством в отличие от прочих свойств и признаков, являющихся откликами на внешние воздействия, проявляющихся и формирующихся во взаимодействии со средой. Именно конституция минерала определяет его видовую принадлежность, диагностические же свойства (признаки) минералов, являющиеся функцией его состава и структуры, служат для установления видовой принадлежности. Поэтому все современные определения понятия минерального вида и классификации минералов основаны не на признаках минералов, а на их конституции. Ниже мы рассмотрим основы конституции минералов более подробно.
В связи с этим вспомним некоторые важнейшие для нас положения физики, химии и кристаллохимии.
Агрегатное состояние минералов. Любое вещество неорганической природы в зависимости от температуры и давления может находиться в любом агрегатном состоянии, а при изменении этих факторов переходит из одного состояния в другое. Минералы, являясь кристаллическими телами по определению, относятся к веществу, находящемуся в конденсированном состоянии, более точно — к твердым телам. Лишь малая часть объектов минералогии принадлежит к числу аморфных тел.
Пределы устойчивости каждого агрегатного состояния находятся в самых различных температурных интервалах в зависимости от природы вещества. При атмосферном давлении в условиях комнатной температуры большинство минералов находятся в твердом состоянии и плавятся при высоких температурах, тогда как ртуть, подобно воде, в этих условиях существует в жидком виде и минералом считаться не может. Однако нетрудно представить себе природные условия, которые позволяют ртути, а тем более воде находиться в кристаллическом состоянии и быть минералами.
Абсолютное большинство минералов представлены кристаллическими веществами, т. е. веществами, обладающими кристаллической структурой. Каждое кристаллическое вещество имеет определенную температуру плавления, при которой изменение агрегатного состояния вещества происходит с поглощением тепла, что ясно сказывается на поведении кривых нагревания (рис. 5а). На некотором интервале времени сообщаемое системе тепло расходуется на процесс плавления (кривая выполаживается).
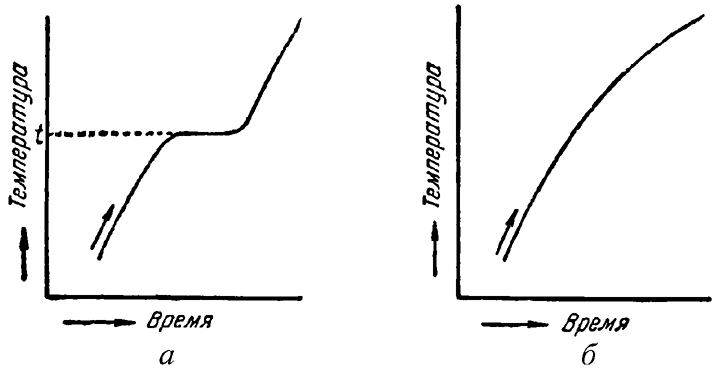
Рис. 5. Кривые нагревания кристаллического (а) и аморфного (б) веществ
Кристаллизация охлаждаемого гомогенного жидкого вещества должна происходить при той же температуре, что и плавление твердого тела того же состава, но обычно она наступает при некотором переохлаждении жидкости, что всегда необходимо иметь в виду.
Твердые химически однородные вещества, характеризующиеся беспорядочной структурой, т. е. отсутствием закономерного расположения атомов, носят название аморфных (стеклообразных) тел. Они принадлежат к числу изотропных веществ, т. е. обладающих по всем направлениям одинаковыми физическими свойствами. Характерной особенностью аморфных веществ, в отличие от кристаллических, является также постепенный переход одного агрегатного состояния в другое по плавной кривой (рис. 5б) подобно сургучу, который при нагревании постепенно становится гибким, затем вязким и, наконец, капельно-жидким. Аморфные вещества, такие как стекло, можно считать переохлажденными жидкостями.
Аморфные вещества часто получаются при затвердевании расплавленных вязких масс, особенно когда охлаждение расплава происходит очень быстро. Примером может служить образование лешательерита — аморфного кварцевого стекла — при ударе молнии в кварцевые кристаллические породы. Переход аморфных веществ в кристаллические массы может произойти лишь при продолжительном выдерживании их в размягченном состоянии при температуре, близкой к точке плавления.
О кристаллическом строении вещества. Кристаллическое состояние определяется наличием в конденсированном теле ближнего и дальнего порядка в расположении атомов, что можно кратко выразить термином «решетчатое строение». Необходимо помнить, что ни кристалл, ни кристаллическая структура сами по себе не тождественны решетке, которая является скорее геометрическим образом, описывающим параллельно-переносную (трансляционную) симметрию трехмерно-регулярной атомной постройки. Вероятно, наиболее корректным использованием понятия кристаллической решетки в отношении кристалла является образное выражение акад. Н. В. Белова: «Кристалл находится в состоянии решетки».
Полная симметрия любой кристаллической структуры описывается одной из 230 пространственных (федоровских) групп симметрии, которые предопределяют одну из 32 групп симметрии (точечные группы, или виды симметрии) естественных многогранников, которые могут образовываться при кристаллизации вещества с данной структурой.
Надежными признаками кристаллического состояния веществ, кроме характера кривых плавления, являются анизотропия физических свойств кристаллических индивидов и дифракция рентгеновских лучей, которая может наблюдаться даже в отношении поликристаллических масс, в том числе и порошка.
Напомним, что строение кристаллического вещества определяется: 1) относительным числом структурных единиц (атомов, ионов, молекул), удерживаемых в пространстве в упорядоченном состоянии электростатическими силами; 2) соотношением размеров структурных единиц, с чем связаны плотность упаковки и координационное число (т. е. число ближайших анионов, окружающих данный катион); 3) их химическими связями, что также играет существенную роль в пространственном расположении атомов или ионов с образованием различных типов структур; 4) термодинамическими параметрами (температура и давление), при которых вещество существует.
Силы связей, которыми структурные единицы в кристаллах удерживаются в равновесии, в различных типах химических соединений неодинаковы по своей природе. Для подавляющего большинства неорганических кристаллических веществ типична ионная связь, характеризующаяся тем, что силы связи обусловлены электростатическим притяжением противоположно заряженных ионов (например, Na1+ и Сl1– в кристаллической структуре NaCl). Для многих кристаллических веществ устанавливается направленная ковалентная (гомополярная) связь, выражающаяся в том, что тесно сближенные атомы для образования устойчивых наружных электронных оболочек одну или несколько пар электронов используют совместно (например, в структуре алмаза каждый атом, прочно связанный с четырьмя окружающими атомами, образует четыре ковалентные связи). В кристаллических структурах металлов распространена металлическая связь, обусловленная тем, что «избыточные» в наружной электронной оболочке атомов электроны не теряются, а образуют общий «электронный газ» вокруг положительно заряженного остова структурных единиц. В молекулярных структурах структурные единицы, представленные электрически нейтральными молекулами, удерживаются слабыми вандерваальсовскими (остаточными) связями (таковы многие органические соединения, а также самородная сера, окись сурьмы и др.). Кроме того, существуют кристаллические вещества, в которых одновременно устанавливаются разные типы связей с преобладанием одной из них. Особенностями связи структурных единиц обусловлены многие свойства минералов (оптические, механические, электропроводность, теплопроводность и др.).
В ионных соединениях анионы, как относительно крупные структурные единицы, занимают главное пространство, а в кристаллических структурах и при плотной упаковке, естественно, стремятся к правильному расположению в пространстве по закону кубической (трехслойной) или гексагональной (двухслойной) плотнейших упаковок. Катионы же ввиду их меньших размеров размещаются в промежутках между анионами — в тетраэдрических и октаэдрических «пустотах» в зависимости от их относительных размеров. Как известно, число октаэдрических пустот в средах с плотнейшей упаковкой равно числу анионов, а число меньших по размерам тетраэдрических пустот в два раза больше. Однако не все эти пустоты обязательно заполняются катионами, причем заполнение может происходить разными способами: рядами, слоями, кольцами, зигзагообразно и т. п. Тетраэдрической и октаэдрической формой пустот в значительной мере обусловлен тот факт, что координационными числами катионов являются по большей части 4 и 6.
Теория плотнейших упаковок для неорганических соединений подробно разработана Н. В. Беловым и весьма плодотворно применена к расшифровке сложных кристаллических структур многих минералов с выявлением важнейших структурных деталей, обусловливающих те или иные свойства кристаллических веществ. Однако следует иметь в виду, что в зависимости от размеров катионов существуют и менее плотные упаковки (с «раздвинутыми» анионами), и такие, которые не могут быть причислены к плотнейшим (например, полевые шпаты).
Кристаллизация вещества, как известно, является экзотермическим процессом, т. е. совершается с выделением тепла. Энергии кристаллических веществ ионных соединений, не содержащих сильно поляризующих или поляризуемых ионов, как показал А. Ф. Капустинский, увеличиваются с увеличением числа структурных единиц (ионов) и их степеней окисления, а также с уменьшением их размеров (радиусов ионов). Энергией кристаллического вещества обусловлены такие свойства, как растворимость, летучесть, температура плавления, до некоторой степени твердость и другие свойства, характеризующие устойчивость соединения.
Полиморфизм и политипия. Полиморфизмом (от греч. поли — много, морфэ — форма) называют способность данного кристаллического вещества при изменении внешних факторов (главным образом температуры) претерпевать одно или несколько видоизменений кристаллической структуры, а в связи с этим и изменений физических свойств. Такие превращения называются полиморфными переходами; они являются фазовыми переходами в твердом состоянии.
Наиболее ярким примером в этом отношении является диморфизм природного углерода, кристаллизующегося в зависимости от условий либо в виде алмаза (кубическая сингония), либо в виде графита (гексагональная сингония), очень сильно отличающихся друг от друга по физическим свойствам, несмотря на тождество состава. При нагревании без доступа кислорода кристаллическая структура алмаза при атмосферном давлении перестраивается в более устойчивую (стабильную) в этих условиях структуру графита. Обратный переход графита в алмаз при атмосферном давлении не устанавливается. Для получения алмаза из графита необходимы давления не менее 25 Кбар. Переход алмаза в графит в поверхностных условиях энергетически обусловлен тем, что алмаз при атмосферном давлении не является устойчивой (т. е. есть стабильной, энергетически выгодной) модификацией ни при каких температурах, однако при умеренных температурах является метастабильным — сохраняется как бы в закаленном состоянии. Переход его в графит энергетически выгоден, но не может начаться при низких температурах по причине большой энергии активации, требующейся для разрушения связей в алмазе. Разогревание обеспечивает эту энергию, и переход осуществляется.
Для многих полиморфных переходов, требующих существенной структурной перестройки (переходы первого рода), часто наблюдается подобная задержка превращения; особенно это характерно для переходов, протекающих на фоне падения температуры. Задержка фазовых переходов первого рода может наблюдаться не только при смене полиморфных модификаций, но даже и при изменении агрегатного состояния вещества — при затвердевании расплава. Длительное существование аморфных тел является одним из примеров метастабильных (закаленных) состояний, когда быстрое охлаждение не позволяет жидкостям кристаллизоваться и они сохраняются в виде стекол.
Иногда полиморфное превращение сопровождается очень незначительным изменением кристаллической структуры вещества (переходы второго рода), и потому без тонких исследований не удается заметить каких-либо существенных изменений в физических свойствах минерала. Таковы, например, превращения так называемого α-кварца и β-кварца и обратно. Однако изучение оптических свойств (рис. 6) однозначно показывает скачкообразное изменение в точке перехода (около 573 °С) таких свойств, как показатели преломления, двупреломление и вращение плоскости оптической поляризации. Полиморфные переходы второго рода обычно не испытывают задержки, за исключением так называемых непрерывных переходов типа порядок-беспорядок, которые будут рассмотрены ниже при обсуждении изоморфизма.
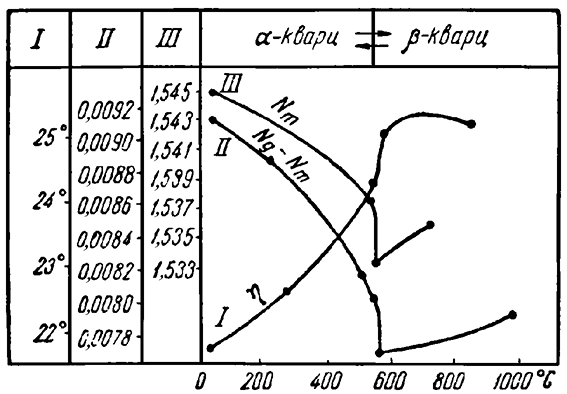
Рис. 6. Изменение свойств кварца при нагревании.
I — вращение плоскости поляризации; II — величина двупреломления;
III — показатель преломления Nm (для линии Dспектра)
Устойчивые в тех или иных определенных физико-химических условиях разности данного кристаллического вещества называются полиморфными модификациями, каждая из которых характеризуется определенной, ей свойственной кристаллической структурой. Таких полиморфных модификаций у какого-либо конкретного вещества может быть две, три или более (например, для серы установлено шесть модификаций, из которых в природе встречаются только три; для SiO2 — девять модификаций и т. д.). Каждая полиморфная модификация является устойчивой при тех значениях термодинамических параметров (главным образом P и T), при которых она обладает минимальным значением свободной энергии Гиббса (термодинамического потенциала G) среди всех других возможных модификаций. Если достигнуты такие значения термодинамических параметров, при которых две модификации или более имеют равные значения свободной энергии, то при этих условиях две или несколько модификаций находятся в равновесии. Если термодинамические параметры изменяются и проходят через значения, при которых существует равновесие двух модификаций, то одна из них сменяется другой — происходит полиморфный переход. Следовательно, каждая полиморфная модификация характеризуется определенным полем устойчивости на P–T-диаграмме состояния (рис. 7).
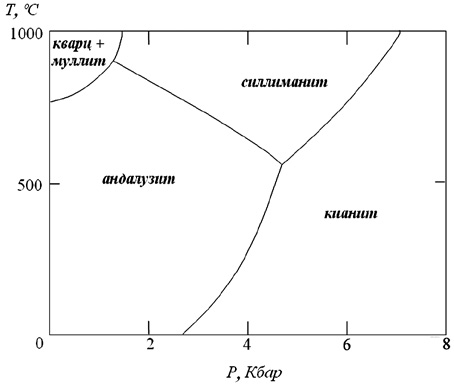
Рис. 7. Полиморфы Al2SiO5 на диаграмме состояния однокомпонентной системы
Различные полиморфные модификации обычно обозначаются приставками к названию минерала греческих букв α, β, γ и т. д. (например: α-кварц, устойчивый при температурах ниже 573 °С; β-кварц, устойчивый при температурах выше 573 °С, и др.). В порядке наименования модификаций в литературе нет единообразия: одни придерживаются обозначения различных модификаций буквами α, β... в порядке повышения или понижения температуры превращения; другие порядок обозначений применяют по степени распространенности или в порядке открытия. Более рациональным следует считать первый порядок обозначения.
Явления полиморфизма весьма широко распространены среди природных соединений. К сожалению, они еще недостаточно изучены. Полиморфные модификации различных минералов могут быть устойчивы в самых различных диапазонах изменения внешних факторов (температуры, давления и др.). Одни обладают широким полем устойчивости при весьма значительных колебаниях температуры и давления (ильменит, графит), другие, наоборот, претерпевают полиморфные превращения в узких пределах изменения внешних факторов (сера).
Понижение температуры при перестройке кристаллической структуры, как правило, приводит к модификации, характеризующейся более высоким координационным числом катиона (например, для нашатыря NH4Cl — 8 вместо 6 при T = 184 °С), что сопровождается уменьшением объема, а следовательно, увеличением плотности (удельного веса) и связанного с нею показателя преломления. Симметрия низкотемпературных модификаций для одного и того же вещества обычно ниже по сравнению с высокотемпературными. Понижение давления, наоборот, должно благоприятствовать уменьшению координационного числа, а стало быть, и обусловливать понижение температуры превращения, в том числе и температуры плавления для огромного большинства веществ, плавящихся с увеличением объема (лед, висмут и сурьма являются исключением из этого правила).
Рост давления приводит к полиморфным переходам с повышением плотности, следовательно, координационное число будет повышаться. В этом отношении действие давления аналогично эффекту понижения температуры, однако симметрия высокобарических модификаций обычно не ниже, а выше симметрии низкобарических. Кроме того, для некоторых соединений большое значение имеет химизм среды: так, сильно щелочные среды при образовании глиноземсодержащих силикатов приводят к уменьшению координационного числа Аl (от 6 до 4).
Для данного типа химических соединений (например, карбонатов) переход от одной кристаллической структуры к другой, как показал В. М. Гольдшмидт, связан с явлением морфотропии, т. е. с преобразованием формы. Морфотропия является не процессом, который происходит во времени, а закономерностью, наблюдаемой в ряду однотипных соединений при фиксированных термодинамических параметрах. Например, в ряду карбонатов Me[CO3] (в скобках — радиусы катионов):
Mg[CO3] (0,74) — Zn[CO3] (0,83) — Са[CO3] (1,04) — Sr[CO3] (l,20) — Ba[CO3] (l,33)
устанавливается, что карбонаты, располагающиеся до Са[CO3] (содержащие меньшие по размерам катионы), кристаллизуются в тригональной сингонии, а после Са[CO3] (с бОльшими размерами катионов) — в ромбической сингонии. Следовательно, существует критическое значение отношения rK : rA, определяющее границу устойчивости двух различных структурных типов. В самом соединении Са[CO3] отношение rK к rA таково, что в зависимости от внешних факторов может образоваться либо кальцит (тригон. с.), либо арагонит (ромбич. с.). Кальцит по сравнению с арагонитом обладает меньшим удельным весом и меньшим показателем преломления. Иначе говоря, карбонат кальция, находящийся на границе морфотропного превращения, является диморфным. Действительно, малые изменения давления и температуры по-разному влияют на величины радиусов катиона и аниона, так что карбонат кальция принимает структуру, отвечающую возникающему отношению rK к rA. Такое существенное сходство полиморфизма с морфотропией позволило В. М. Гольдшмидту называть полиморфизм автоморфотропией.
Если данная модификация кристаллического вещества, допустим α, обладает свойством при изменении внешних условий (например, температуры) переходить в другую — β-модификацию, а при восстановлении прежних условий превращается обратно в α-модификацию, то такие полиморфные превращения называются энантиотропными (от греч. энантиос — противоположный, тропос — перемена, преобразование). Пример: превращение ромбической α-серы в моноклинную β-серу и обратно. Если же обратный переход не может совершаться, то такой вид превращения носит название монотропного. Примером может служить монотропное (необратимое) превращение ромбического арагонита (Ca[CO3]) в тригональный кальцит (при нагревании). Необратимость этого перехода связана с тем, что высокобарический арагонит при нормальном давлении является метастабильным; нагревание вызывает его переход к стабильной модификации и дальнейшее изменение температуры не может превратить его снова в арагонит. Однако если в числе варьируемых параметров, кроме температуры, окажется и давление, переходы в арагонит и обратно станут легкоосуществимыми.
В природе нередко наблюдается одновременное существование в одних и тех же физико-химических условиях двух модификаций даже рядом друг с другом (например, пирита и марказита, кальцита и арагонита и др.). Очевидно, переход одной из модификаций в стабильную, т. е. устойчивую, в силу каких-то причин задержался, и вещество в таком случае находится в метастабильном состоянии, подобно тому как существуют переохлажденные жидкости.
Необходимо отметить, что метастабильные при данных значениях термодинамических параметров вещества могут не только сохраняться в течение достаточно больших промежутков времени, но даже и возникать за счет еще менее энергетически выгодных фаз. Это явление называется метастабильным зарождением и ростом. Так, при выпадении карбоната кальция из водных растворов при температуре ниже 30 °С сначала появляется метастабильный арагонит, который в дальнейшем переходит в кальцит, а иногда может и сохраниться. Такое, казалось бы, парадоксальное поведение карбоната кальция легко объясняется в свете правила ступенчатых переходов Оствальда: переход от наименее стабильной при данных условиях модификации к наиболее стабильной происходит постепенно, через состояния, промежуточные по величине энергии связи. Очевидно, пересыщение водных растворов относительно растворенных компонентов может быть таким сильным, что ставшая нестабильной растворенная форма карбоната кальция по энергии связи отстоит от стабильного кальцита дальше, чем нестабильный арагонит. В такой ситуации переход карбоната из раствора в твердое состояние осуществляется через промежуточную фазу — арагонит.
Следует подчеркнуть, что устойчивая модификация по сравнению с неустойчивой обладает: 1) меньшей упругостью пара; 2) меньшей растворимостью и 3) более высокой температурой плавления. Отметим также, что продукты полиморфных переходов, несмотря на изменение объема, нередко сохраняют внешнюю форму кристаллов ранее существовавшей модификации. Подобные продукты замещения называются параморфозами, примером которых являются октаэдрической формы выделения графита, представляющие параморфозы последнего по алмазу.
К числу явлений, сходных с полиморфизмом, относится особый случай структурного разнообразия веществ одинакового состава, называемый политипией. Различные политипные модификации характерны для веществ со структурой, построенной из одинаковых слоеподобных фрагментов, характер взаимного наложения которых может быть различным за счет, например, различных взаимных разворотов или сдвигов слоев. Различные политипы характеризуются определенной периодичностью в направлении, перпендикулярном к плоскости слоев; зачастую они различаются и симметрией. Политипные модификации обозначаются обычно символами Рамсделла, содержащими стоящие впереди цифры, обозначающие число слоев в периоде, и латинские буквы (Tk, M, O, Q, R, T, H и C для модификаций с ячейками, обладающими триклинной, моноклинной, (орто-)ромбической, тетрагональной, ромбоэдрической, тригональной, гексагональной и кубической симметрией соответственно). После буквенного обозначения может находиться подстрочный числовой индекс, если среди политипных модификаций определенной слойности есть несколько различных с одинаковой симметрией. Для полностью неупорядоченных последовательностей слоев употребляется буквенное обозначение D (от англ. disorder — беспорядок).
Политипия широко распространена среди кристаллических веществ. Так, для самородного серебра, кроме обычной трехслойной кубической модификации 3С, в природе известны двух- и четырехслойная модификации 2H и 4H гексагональной сингонии; у лепидолита могут встречаться две двухслойные моноклинные модификации, обозначаемые 2M1 и 2M2. Особенно богаты политипами, как показал лабораторный синтез, сульфид цинка ZnS (более 150) и карбид кремния SiC (128). Среди синтетических политипов SiC имеется модификация 594R, параметр ячейки вдоль оси С этой модификации составляет около 1500 A o. Два из политипов SiC известны в природе: 3С и 2H. Сульфид цинка ZnS встречается в природе в виде трех политипных модификаций: 3С — сфалерит и вюртциты 2H и 15R. Из природных соединений политипия свойственна также хлоритам (восемь политипов), хегбомиту (семь политипов), слюдам (19 политипов), сульфиду кадмия и молибдениту (по два политипа).
Так как различные политипы одного вещества построены из одинаковых слоев, координационные числа атомов в них не отличаются, что приводит обычно к ничтожным различиям по энергии связи между различными политипами. Благодаря такой близости различных политипов по энергии для них невозможно, за малыми исключениями, выделить индивидуальные поля устойчивости на P–T-диаграммах, следовательно, при одних и тех же условиях могут сосуществовать несколько политипных модификаций. Таким образом, в отличие от полиморфизма в обычных его проявлениях вариации температуры и давления не могут быть признаны главными факторами, определяющими устойчивость различных политипных модификаций. Причины образования тех или иных политипов остаются пока до конца не выясненными. Считается, что на это влияют микропримеси и динамические особенности роста кристаллов. Такой индифферентизм политипных модификаций по отношению к изменению главных термодинамических параметров требует рассматривать политипы особняком от прочих полиморфов, для которых поля устойчивости вполне определенны. Лишь кубические модификации для большинства политипных веществ, являясь наиболее высокосимметричными, тяготеют к максимальным температурам, сменяя все прочие политипы, так что область стабильного существования первых можно указать, хотя и трудно четко ограничить со стороны низких температур.
Явления разрушения кристаллических структур. Главнейшими особенностями кристаллических структур минералов являются закономерное расположение и строго уравновешенное состояние слагающих их структурных единиц. Однако достаточно создать такие условия, при которых внутренние связи структурных единиц будут поколеблены, как из кристаллического вещества с упорядоченной пространственной структурой мы получим аморфную массу, не обладающую кристаллическим строением.
Прекрасным примером в этом отношении является разновидность минерала брусита — ферробрусит: (Mg,Fe) [OH]2, содержащий в виде изоморфной примеси до 36 % (по весу) закиси железа. В свежем состоянии этот минерал, будучи извлечен из глубоких горизонтов шахт, совершенно бесцветен, прозрачен и обладает стеклянным блеском. В течение нескольких дней его кристаллики на воздухе постепенно меняют свой цвет, становясь золотисто-желтыми, затем бурыми и, наконец, непрозрачными темно-коричневыми, сохраняя внешнюю кристаллическую форму1. Химический анализ показывает, что почти все двухвалентное железо при этом переходит в трехвалентное (т. е. происходит окисление), а рентгенометрическое исследование не устанавливает признаков кристаллического строения. Очевидно, окисление железа нарушило внутренние связи в кристаллической структуре, что и привело к дезорганизации строения вещества.
То, что происходит с ферробруситом в окислительной обстановке при комнатной температуре и атмосферном давлении, для других минералов может иметь место при повышенных температурах и давлениях, как это уже установлено для ряда случаев.
Весьма интересные явления изучены в минералах, содержащих редкоземельные и радиоактивные элементы (ортит, фергюсонит, эшинит и др.). В них также очень часто, но не всегда устанавливается превращение кристаллического вещества в аморфное, которое, как предполагают, обязано действию α-лучей при радиоактивном распаде2. Эти измененные стеклообразные минералы, не относящиеся к кубической сингонии, оптически изотропны и не обнаруживают дифракции рентгеновских лучей, т. е. ведут себя как аморфные тела. При этом происходит частичная гидратация вещества. Такие тела Бреггер назвал метамиктными.
В подтверждение явлений распада кристаллических сред можно привести и ряд других аналогичных примеров, иллюстрирующих образование аморфных или коллоидных масс. Однако нельзя думать, что эти новообразования являются устойчивой формой существования вещества. Известно немало примеров вторичной перегруппировки вещества с образованием новых кристаллических тел, устойчивых при изменившихся условиях. Так, известны «кристаллы ильменита» (Fe2+TiO3), которые при микроскопическом изучении оказываются состоящими из смеси двух минералов: гематита (Fe2O3) и рутила (TiO2). По-видимому, после момента образования ильменита в какой-то период жизни минерала под влиянием изменившегося режима кислорода создались резко окислительные условия, приведшие к переходу Fe2+ в Fe3+ с одновременным распадом кристаллической структуры, а затем к постепенной перегруппировке вещества с образованием смеси устойчивых минералов. Точно так же, например, наблюдались случаи образования на месте тиллита (PbSnS2), галенита (PbS) и касситерита (SnO2) в теснейшем прорастании друг с другом, но при сохранении реликтового (т. е. прежнего) пластинчато-зернистого строения агрегата, характерного для тиллита. Очевидно, в связи с увеличившейся в какой-то момент концентрацией кислорода в данной среде олово, обладая большим сродством с кислородом, обособилось из первоначально гомогенной минеральной массы в виде окисла, а свинец перешел в форму самостоятельного сернистого соединения.
Минералы, находящиеся в метамиктном состоянии, как метастабильные фазы, имеют некоторый запас потенциальной энергии по сравнению с устойчивой модификацией. Эта энергия связана со смещением атомов из равновесных положений, произошедшим при столкновении с осколками деления; при низких температурах амплитуды тепловых движений атомов недостаточны для их возвращения в положения, которые были характерны для структуры до начала метамиктного распада. Нагревание метамиктных минералов чаще всего позволяет вывести их из метастабильного состояния с восстановлением первоначального кристаллического состояния. Нередко восстановление структуры происходит за столь короткое время, что бурно выделяющийся избыток энергии, отвечающий теплоте плавления, приводит к почти мгновенному самопроизвольному разогреву минерала до высокой температуры, сопровождаемому растрескиванием и интенсивным свечением (так называемая рекалесценция).
2.2. Химический состав и формулы минералов
Выше мы уже упоминали о том, что подавляющее большинство встречающихся в природе минералов представлено химическими соединениями. Среди последних различают: а) соединения постоянного состава (дальтониды) и б) соединения переменного состава.
Соединения постоянного состава. Все химические соединения постоянного состава, как известно, строго подчиняются закону кратных отношений (закон Дальтона) и закону валентных паев, связывающему отношения компонентов данного соединения с отношениями их в других типах соединений. Эти законы находятся в полном соответствии с периодической системой элементов Д. И. Менделеева, законами кристаллохимии и учением о симметрии в кристаллических средах.
Характерно, что эти соединения отличаются целым рядом особых физических свойств, отчетливо выступающих на физико-химических диаграммах плавкости, растворимости, электропроводности, твердости, удельного веса, показателей преломления и пр.
Необходимо отметить, что реальные минералы в отношении своего химизма практически никогда не могут рассматриваться в качестве соединений постоянного состава, поскольку содержат то или иное изменяющееся от индивида к индивиду количество примесей (минеральные индивиды также можно считать химически однородными лишь в первом приближении). Причина такого положения дел заключается, во-первых, в одном из качеств природных минералообразующих систем, которое определяется законом В. И. Вернадского о «всюдности химических элементов». Второй причиной является чрезвычайная энергетическая выгодность наличия примесей в кристаллической структуре по сравнению с чистым веществом. Дело в том, что добавление малого количества примеси (не механической, а в атомной форме) вызывает резкое повышение так называемой конфигурационной энтропии соединения по сравнению с нулевым значением этого параметра для чистого вещества, что оказывает стабилизирующее воздействие на кристаллическую структуру, тем большее, чем выше температура. Всякое введение посторонних частиц в структуру, конечно, стремится дестабилизировать ее, вызывая в ней напряжения, но для малых количеств примеси энтропийный положительный эффект всегда превышает потери, затрачиваемые на «содержание» примеси. Поэтому ни одно природное соединение без особых причин не может «отказаться» от присутствия хотя бы небольших порций посторонних атомов в структуре, тем более когда состав минералообразующей системы обеспечивает широкий выбор наиболее подходящих примесных атомов.
Лишь отдельные индивиды минералов, формирующиеся в специфических условиях природной дистилляции, приближаются к соединениям постоянного состава. Тем не менее в качестве идеализации многие минералы полезно условно рассматривать как соединения постоянного состава. Такое абстрагирование от реального состава позволяет использовать понятие минерального вида и характеризовать индивиды минералов, относящиеся к определенному минеральному виду, единообразными и относительно простыми химическими формулами, отражающими устойчивые особенности их, вообще говоря, непостоянного состава.
К числу бинарных химических соединений относятся: простые оксиды (Cu2O, MgO, Fe2O3, SiO2 и др.), сульфиды (NiS, FeS2, Sb2S3 и др.), галогениды (NaCl, AgBr, CaF2 и др.) и т. д. Соединения, состоящие из атомов трех элементов, называются тернарными; таковы различные кислородные соли (Са[CO3], Ca[SO4], Y[PO4], Mg2[SiO4] и др.). Существуют, конечно, и соединения более сложного состава.
Химический состав химических соединений может изображаться двояким способом: 1) в виде эмпирических формул; 2) в виде конституционных или структурных формул.
Эмпирическими формулами выражают состав минералов либо в виде символов элементов, входящих в соединение, начиная с наиболее низковалентных катионов и заканчивая наиболее низковалентными анионами (например, Ba[SO4], Na3[AlF6], Na[AlSi3O8] и т. д.), либо в виде ряда простейших составляющих соединений (BaO . SO3, 3NaF . AlF3, Na2О . Al2O3. . 6SiO2 и т. д.). Последний вид формул хотя и не отвечает современным представлениям о природе химических соединений, однако имеет то преимущество, что позволяет легче запомнить состав минералов и в удобной форме записывать уравнения реакций с их участием.
Существенные поправки в начертание формул более сложных соединений вносят данные кристаллохимии, основывающиеся на рентгеноструктурном изучении минералов. Так как большинство неорганических кристаллических веществ характеризуются ионными связями структурных единиц, то в сложных формулах химических соединений важно отражать эти структурные элементы (катионы и анионные комплексы), устанавливаемые в различных типах кристаллических структур. Анионные комплексы обычно отделяются от катионов квадратными скобками, гидроксильная группа — круглыми, например Са[СО3], Ba[SO4], Na[AlSi3O8], Mg3[Si4O10](OH)2 и т. п. Нередко в подобных формулах дополнительно отражается топология анионных комплексов, для чего после скобок располагаются в виде верхних индексов несколько знаков ∞; их количество соответствует числу измерений, в которых простирается анионная группировка. Так, наличие цепочечного (одномерного) кремнекислородного радикала в структуре минерала группы пироксенов — диопсида — передается в его формуле следующим образом: CaMg [Si2O6]∞. Присутствие слоевых (двумерных) радикалов в структуре талька отражается в его формуле так: Mg3[Si4O10]∞ ∞ (OH)2.
Во многих случаях по таким кристаллохимическим формулам мы можем заранее догадываться о тех или иных физических свойствах соединения. Известно, например, что оптические свойства (в частности, двупреломление) часто зависят от формы анионного комплекса и его ориентировки. Наличие в кристаллической структуре параллельно ориентированных анионов плоской формы [СO3]2– или [Si2O5]2– обусловливает высокое двупреломление (сильно выраженную оптическую анизотропию при отрицательном оптическом знаке). Если же анион представлен группой изометрической формы [SO4]2– или каркасом [AlSi3O8]1–, развитым в пространстве более или менее равномерно, то такие минералы обычно обладают низким двупреломлением. Форма комплексных анионов нередко отражается и на облике кристаллов: слюды, характеризующиеся непрерывно вытянутыми в двух направлениях анионными листами, зачастую характеризуются пластинчатыми формами кристаллов, а пироксены, в которых анионы представлены непрерывно вытянутыми в одном направлении цепочками, обладают призматическим обликом кристаллов и т. д.
Помимо бинарных и тернарных химических соединений, в природе распространены и более сложные соединения, например двойные соли. Двойными солями называются такие соединения постоянного состава, которые состоят как бы из двух простых солей, присутствующих в кратных отношениях. В большинстве случаев эти соли являются двойными по катионам, реже — по анионам или одновременно по катионам и анионам. В качестве примеров могут быть приведены следующие: CaMg[CO3]2, K3Na[SO4]2 и т. д.
При сравнении формул двойных солей выясняется, что входящие в их состав катионы, благодаря значительной разнице их ионных радиусов, не могут изоморфно замещать друг друга: Са2+(1,04 A o) и Mg2+(0,74 A o), К1+(1,33 A o) и Na1+(0,98 A o) и т. д. Поэтому неудивительно, что от составляющих их простых солей двойные соли отличаются некоторыми особенностями кристаллических структур и физическими свойствами.
Соединения переменного состава (твердые растворы, смешанные кристаллы, изоморфные смеси). Кроме химических соединений постоянного состава, получаемых обычно в лабораториях с использованием чистых исходных компонентов, существует огромное множество таких соединений, состав которых не является постоянным, а колеблется то в узких, то в более широких пределах, причем эти колебания состава не могут быть объяснены наличием каких-либо механических примесей посторонних веществ. Наоборот, колеблющийся состав соединений с кристаллохимической точки зрения находит объяснение в растворимости составных компонентов в данном соединении. Такие химические образования получили название соединений переменного состава.
Среди минералов соединения переменного состава составляют большинство. Существование веществ (фаз) переменного состава кажется вполне естественным, если речь идет о жидких растворах, состав которых определяется соотношением количеств растворенного вещества и растворителя. Таким образом, состав раствора колеблется в пределах от чистого растворителя до насыщенного раствора; он может быть любым из непрерывного множества возможных составов в пределах, зависящих от температуры и давления. Способность кристаллических веществ различного состава образовывать непрерывно меняющиеся по составу соединения одинаковой кристаллической структуры основана на изоморфизме, т. е. свойстве атомов различных элементов заменять друг друга в твердых химических соединениях.
Понятие изоморфизма приложимо как к атомам различных элементов, способным выполнять в структуре кристаллов одинаковую кристаллохимическую функцию, статистически размещаясь в позициях одного типа, так и к кристаллам чистых веществ, которые могут образовывать промежуточные произвольные по составу соединения — твердые растворы — благодаря изоморфизму составляющих их атомов. Изоморфизм, подобно морфотропии, не должен восприниматься как процесс или обменная реакция, хотя соединение переменного состава и удобно описывать как продукт реакции замещения или растворения в твердом состоянии. На самом деле твердые растворы в природе в подавляющем большинстве случаев сразу образуются с максимально допустимым при данных условиях количеством изоморфных примесей, так что дальнейшие процессы могут протекать лишь в сторону распада раствора и избавления от примесей на фоне падения температуры. Исключение составляют минералы, находящиеся в условиях нарастающей температуры при прогрессивном метаморфизме.
Область возможных составов твердых растворов, допустимых при фиксированных значениях термодинамических параметров, вместе с составами чистых компонентов образует так называемый изоморфный ряд. Со стороны крайних членов изоморфных рядов, представляющих чистые компоненты, область допустимых составов всегда имеет непрерывный характер, в областях же промежуточных составов может наблюдаться область несмесимости; в таких случаях говорят о существовании разрыва смесимости в ряду твердых растворов. При отсутствии области несмесимости изоморфизм называется совершенным, в противном случае речь идет об ограниченной смесимости и изоморфизм этого типа называется несовершенным.
В физической химии давно уже было установлено, что степень смесимости компонентов зависит от внешних факторов, главным образом от температуры: в условиях высоких температур изоморфное замещение компонентов происходит в гораздо более широких пределах, чем при низких температурах. Следовательно, при повышении температуры изоморфизм в некоторых рядах может из несовершенного сделаться совершенным. Тем не менее некоторые компоненты имеют настолько низкую смесимость в твердом состоянии, что повышение температуры не может сделать крайне ограниченный, несовершенный изоморфизм в таких системах совершенным — один или оба чистых компонента плавятся или разлагаются гораздо раньше, чем исчезнет разрыв смесимости.
В минералогии на зависимость изоморфизма от термодинамических условий впервые обратил внимание В. И. Вернадский в 1910 г. Он указал на существование более широких изоморфных рядов химических элементов для области глубинных магматических образований (высокие P и T) по сравнению с областью метаморфических пород в литосфере (высокое P и средняя T) и с областью выветривания (низкие P и T).
В настоящее время представления об изоморфизме на основе достижений кристаллохимии сильно расширились. Явление изоморфизма имеет несколько взаимосвязанных аспектов, определяющих его многообразие.
Во-первых, можно рассматривать изоморфизм в отношении структурного состояния крайних членов изоморфного ряда. Как предполагалось во время, следовавшее непосредственно после открытия явления изоморфизма, для осуществления изоморфной смесимости в широких пределах необходима изоструктурность чистых компонентов, т. е. крайние члены изоморфных рядов должны обладать близкими структурами — относиться к одному структурному типу. Такое требование и отображено в самом термине изоморфизм, означающем по-гречески равенство формы. Позже выяснилось, что изоморфная смесимость может наблюдаться и между веществами с различными структурными типами. Подобное явление получило название изодиморфизм. Примером системы с изодиморфизмом является непрерывный ряд от кубического Ir до гексагонального Os. Смена структурного типа твердого раствора, которую можно рассматривать в качестве морфотропного перехода, осуществляется «скачком» при составе приблизительно (Ir0,75Os0,25).
Дальнейшие различия в характере изоморфных замещений могут быть обнаружены при анализе общего числа атомов (или других структурных единиц) в крайних членах изоморфного ряда и в твердом растворе.
Наиболее простым и интуитивно ясным является замещение типа «атом на атом» или «молекула на молекулу» («радикал на радикал» в случае изоморфизма по анионной части). Такое замещение происходит с сохранением общего числа атомов (молекул) в системе, а так как при изоморфизме должна сохраняться электронейтральность, то это требует равенства валентностей, точнее, зарядов замещающих друг друга структурных единиц. Изовалентный изоморфизм, характеризующийся заменой в кристаллической структуре ионов одинаковой валентности, широко проявляется при условии, если свойства и размеры взаимно замещающих ионов близки друг к другу (разность ионных радиусов не превышает 15 % от меньшего радиуса согласно правилу Гольдшмидта). Таковы, например, двухвалентные катионы (в скобках указаны размеры ионных радиусов): Mg2+(0,74), Fe2+(0,80), Ni2+(0,74), Zn2+(0,83), Mn2+(0,91) и др.; трехвалентные катионы: Fe3+(0,64), Cr3+(0,64), Al3+(0,57) и др. То же самое относится и к анионам, участвующим в строении кристаллических веществ: S2–(1,82), Se2–(l,93) и др. Из простейших соединений, характеризующихся совершенным изоморфизмом, приведем следующие ряды: MgCO3 — FeCO3, CuS — CuSe и др.
Гетеровалентный изоморфизм отличается тем, что в кристаллических структурах происходит замена одного иона примерно равновеликим ионом иной валентности, однако при условии компенсации зарядов в какой-либо другой паре ионов, участвующих в кристаллическом строении данного вещества, но существенно отличающихся по своим размерам от предыдущих. Таков, например, известный изоморфизм в ряду плагиоклазов: Na[AlSi3O8] — Ca[Al2Si2O8]. Здесь Na1+(0,98) заменяется большим по валентности Са2+(1,04) при одновременной замене одного иона Si4+(0,39) на меньший по валентности Аl3+(0,57). Гетеровалентный изоморфизм в плагиоклазах может быть описан следующей схемой: Ca2+Al3+ → Na+Si4+. В некоторых рутилах (TiO2) наблюдается одновременное присутствие примеси Nb5+ или Ta5+ с Fe3+ (ильменорутил или стрюверит соответственно); гетеровалентное замещение происходит в них по следующей схеме: Me5+Fe3+ → 2Ti4+. Таким образом, общий электростатический баланс соединения сохраняется. Описанные случаи гетеровалентного изоморфизма также относятся к изоморфизму с сохранением числа атомов в системе.
При гетеровалентном изоморфизме кроме компенсации заряда решающую роль играют все же размеры заменяющих друг друга структурных единиц — катионов или анионов (они должны быть более или менее равновеликими).
Однако число структурных единиц при замещении не обязательно должно сохраняться. Например, в слюдах на месте трех двухвалентных катионов Mg (в шестерной координации) могут располагаться два трехвалентных катиона Al (3Mg замещаются на 2Al). Третье место остается вакантным. В схемы подобных замещений вводится символ незанятой позиции, называемой вакансией, — . Для описанного замещения в слюдах схема изоморфизма такова: 2Al3+ → 3Mg2+.
Если рассматривать слюды с подобными изоморфными замещениями в направлении от магнезиальных членов ряда к глиноземистым, мы увидим, что общее число атомов уменьшается. Такой изоморфизм называется изоморфизмом вычитания, или дефектным изоморфизмом. С другой стороны, можно рассматривать слюды этого ряда и в обратном направлении, тогда речь будет идти об изоморфизме заполнения (или внедрения), так как в этом случае мы будем переходить к структурам со все большей степенью заполнения вакансий катионами Mg2+. Ясно, что изоморфизм с участием вакансий приобретает характер вычитания или заполнения в зависимости от способа рассмотрения изоморфного ряда. Определенность здесь может возникнуть, лишь если один из крайних членов подобного твердого раствора вообще не существует в чистом виде. В таком случае мы вынуждены говорить о дефектной (или заполненной) структуре как о некоторой химической модификации, отклоняющейся по составу от стехиометрического состава чистого крайнего члена в направлении недостижимого (гипотетического) соединения. Для большинства же систем с изоморфной смесимостью любого типа, в которых оба крайних члена существуют, особенно при совершенном изоморфизме, допустимо в схемах изоморфных замещений применять двустороннюю стрелку вместо односторонней.
Из вышесказанного ясно, что системы с изоморфными замещениями могут быть и более чем двухкомпонентными. В случае таких систем речь уже идет не о рядах, а о полях и целых областях изоморфной смесимости, как это характерно, например, для полевых шпатов (рис. 8) при высоких температурах, когда смешению подвергаются плагиоклазы и калиевые полевые шпаты.
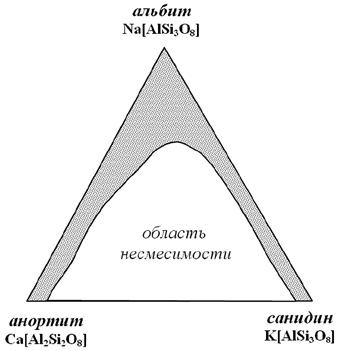
Рис. 8. Поле составов твердых растворов высокотемпературных
полевых шпатов (выделено крапом)
Как мы знаем, ионные радиусы в вертикальных группах периодической системы элементов возрастают с увеличением порядкового номера и уменьшаются в горизонтальном направлении с увеличением номера группы (т. е. с увеличением валентности). На этом основании А. Е. Ферсманом выведен закон диагональных рядов изоморфных ионов в периодической системе элементов, справедливый для левой ее части. Намечаются следующие гетеровалентные ряды изоморфизма ионов (в скобках показаны ионные радиусы в ангстремах):
Li1+(0,68) — Mg2+(0,74) — Sc3+(0,83) — Zr4+(0,82);
Na1+(0,98) — Са2+(1,04) — Y3+(0,97);
K1+(1,33) — Sr2+(1,20);
Rbl+(l,49) — Ba2+(1,38);
Al3+(0,57) — Ti4+(0,64) — Nb5+(0,66).
Действительно, в природных соединениях мы нередко наблюдаем, что литиевые минералы, например, содержат изоморфные примеси магния, магниевые минералы — примеси скандия, натриевые — примеси кальция, кальциевые — примеси иттрия и т. д.
Кроме того, в комплексных анионах ион [SiO4]4– может заменяться ионами [АlO4]5–, [РО4]3– и [SO4]2–, имеющими одинаковые или близкие размеры. На приведенных примерах мы не раз будем останавливаться при описании минералов.
Исследования А. Е. Ферсмана и В. М. Гольдшмидта показали, что влияние размерных характеристик атомов на возможность и пределы изоморфных замещений оказывается несимметричным в отношении бо/льших и меньших ионов и связано также с их зарядами (валентностью). Это выражено правилом полярности изоморфизма: высокозарядные малые ионы легче (в большем количестве) входят в структуру, чем ионы большего радиуса и с меньшим зарядом.
В настоящее время, во многом благодаря исследованиям акад. В. С. Урусова, можно считать установленными основные закономерности изоморфизма, многие из которых получили объяснение в рамках энергетической кристаллохимии. Так, выяснено, что весьма существенным фактором изоморфных замещений является близость электронных характеристик замещающих друг друга атомов, наиболее просто выражаемая через такие их параметры, как электроотрицательность. Помимо размерного фактора, учитываемого правилом Гольдшмидта об отличии в радиусах ионов не более 15 %, для обеспечения изоморфной смесимости требуется и близость величин их электроотрицательности. Кроме того, для сложных по составу соединений в которых влияние геометрических параметров замещающих друг друга атомов благодаря малости доли занимаемого ими общего объема в структуре, слабо сказывается на параметрах решетки, требования не более чем 15 % различия радиусов могут быть существенно смягчены. Установлен также характер влияния повышенного давления на изоморфизм: пределы изоморфной смесимости в целом сужаются, однако энергетически выгодным может являться вхождение катиона в структуру минерала, если оно приведет к увеличению координационного числа атомов примеси. Этот эффект иллюстрируется высокой глиноземистостью пироксенов и амфиболов глубоко метаморфизованных пород за счет повышенных содержаний четырехкоординированного алюминия.
Среди природных соединений переменного состава с генетической и кристаллохимической точки зрения важно различать два типа: 1) истинные твердые растворы; 2) микронеоднородные минералы (в том числе твердые псевдорастворы, многие из которых представляют собой продукты распада твердых растворов).
Истинные твердые растворы, или, как их иначе называют, изоморфные смеси, представляют собой совершенно однородные по кристаллической структуре смеси в любых пропорциях двух или нескольких веществ, не образующих химических соединений промежуточного состава. Примерами могут служить твердые растворы золота и серебра, оливины, закаленные плагиоклазы вулканического происхождения и т. д.
Физические и химические свойства твердых растворов являются аддитивными (от лат. addo — прибавляю), т. е. постепенно и закономерно изменяющимися при увеличении содержания второго компонента. Таковы, например, изменения температуры плавления, удельного веса, показателей преломления, отражательной способности, электропроводности и т. д. Изменения всех этих свойств на диаграммах выражаются в виде практически прямых линий (для удельных весов и параметров решетки) или плавно изгибающихся кривых (температур плавления, оптических свойств и др.). Эти кривые для изоморфных рядов настолько характерны, что по совокупности изученных свойств какого-либо минерального вида переменного состава по ним можно определить состав конкретного индивида, измерив значения его свойств и не прибегая к химическому анализу. Например, нетрудно определить таким путем по специальным диаграммам состав оливина, изучив его оптические свойства в шлифах под микроскопом.
В химических формулах твердых растворов изоморфные атомы или ионы ставят в круглые скобки, отделяя друг от друга запятыми и располагая в убывающем порядке содержания: (Au,Ag), (Zn,Fe)S, (Zn,Fe)CO3, (Fe,Mn)WO4 и т. д. Если в двойной соли один из компонентов содержит изоморфные примеси, то формула изображается в следующем виде: Ca(Mg,Fe)[CO3]2.
Приведенные выше формулы твердых растворов, выражающие химизм минералов переменного состава в обобщенном виде, содержат информацию о соотношении изоморфных атомов лишь в порядковой форме (больше — меньше). Химический состав конкретных минеральных индивидов, оцениваемый по результатам их химического анализа, выражается в так называемых эмпирических формулах, например, состав одного из клинопироксенов Хибинского массива описывается следующей эмпирической формулой: (Na0,88Ca0,10K0,02)1,00(Fe3+0,88Mg0,05Al0,03Fe2+0,02Ti4+0,02)1,00 [(Si1,95Al0,05)2,00O6].
Микронеоднородными минералами считаются такие минеральные индивиды, неоднородность которых устанавливается лишь при микроскопических исследованиях. Внешне кристаллы подобных минералов могут казаться однородными, однако при микроскопических, а иногда — лишь при электронно-микроскопических исследованиях такие минералы обнаруживают неоднородное строение. Неоднородности могут быть представлены как минеральными включениями различного масштаба, так и многофазными включениями, представляющими фрагменты минералообразующей среды (расплавные, газово-жидкие включения).
Во многих случаях неоднородность минералов является первичной, формируясь непосредственно в процессе их образования в результате захвата посторонних минеральных частиц и отдельных порций минералообразующей среды через поверхность растущего кристалла. В целом ряде случаев минеральные микровключения занимают ориентированное положение относительно кристалла-хозяина. Иногда такие включения возникают при захвате ориентированно наросших (эпитаксических) микроиндивидов других минералов (например, тончайшие пластины ильменита или слюды, ориентированные параллельно граням базопинакоида кристаллов корунда). Если ориентированно наросшие минералы впоследствии оказываются заключенными внутри кристалла-хозяина, образующиеся вростки называются эндотаксическими. Нередко микроиндивиды постороннего минерала, имеющие закономерную ориентировку относительно кристалла-хозяина, росли одновременно с ним (топотаксия).
Интересный пример неоднородности представляют синтаксические срастания структурно родственных минералов, образующих отдельные блоки, представленные в различных объемных отношениях. Подобное явление на ультрамикроскопическом уровне наблюдается в минералах, представленных монокристаллами, в строении которых участвуют различные по составу и строению, но хорошо сочетающиеся между собой регулярно расположенные структурные блоки, присутствующие в кратных отношениях. Минералы, построенные таким образом, называются полисомами и образуют дискретные ряды — полисоматические серии, например минералы серии бастнезит-паризит. Если полисоматические структуры сложены плоскими блоками (подобно политипам — слоями, но различными по составу), возникают так называемые смешаннослойные образования, к которым, как показали электронно-микроскопические исследования, относятся многие глинистые минералы, ранее считавшиеся однородными.
Всевозможные примеси и дефекты в кристаллах минералов, обладая сами по себе микроскопическими размерами, могут неравномерно распределяться в минералах, делая их неоднородными и для невооруженного взгляда. Как изоморфные, так и механические примеси нередко в различных количествах захватываются растущим кристаллом на различных этапах его роста, что приводит к зональному распределению окраски; подобные зоны могут наблюдаться и макроскопически, формируя ритмично-полосчатую зональность. Если примеси неодинаковым образом поступают в минеральный индивид через различные элементы огранения растущего кристалла (вершины, ребра, различные грани), распределение примесей может иметь секториальный характер.
Весьма распространены в реальных минеральных индивидах и случаи вторичной неоднородности, по большей части возникающей в процессе низкотемпературной эволюции минералов, представлявших при своем возникновении истинные твердые растворы. Так как большинство минеральных индивидов после своего образования претерпевают охлаждение, пределы изоморфной смесимости сужаются и твердый раствор начинает испытывать превращения, сводящиеся часто к распаду (экссолюции) с выделением избыточных компонентов в виде новых минеральных фаз. Например, в экспериментально изученной системе NaCl — KСl эти соединения при высоких температурах образуют непрерывный ряд твердых растворов. Однако при постепенном охлаждении до комнатной температуры в закристаллизовавшейся массе возникают мельчайшие закономерно сросшиеся тельца NaCl и KСl. Количественные их соотношения зависят от исходного состава твердого раствора.
Теоретический состав минеральных фаз, которые должны образоваться при распаде твердого раствора, определяется на диаграмме «состав —температура» (рис. 9) положением кривой равновесного сольвуса (бинодали). В большинстве случаев образующиеся фазы зарождаются при более низких температурах, чем те, которые задаются равновесным сольвусом. Зарождение индивидов новой фазы в твердом теле требует дополнительной энергии на преодоление упругих деформаций при согласовании поверхностей выделяющейся фазы и кристалла-матрицы, что достигается переохлаждением по сравнению с температурой теоретического «насыщения»; реальный состав выделяющейся фазы и кристалла-матрицы определяется линией когерентного сольвуса. При длительном глубоком переохлаждении структура распада делается более грубой и когерентность может нарушиться, после чего состав сосуществующих фаз достигает величин, задаваемых равновесным сольвусом (см. рис. 9).
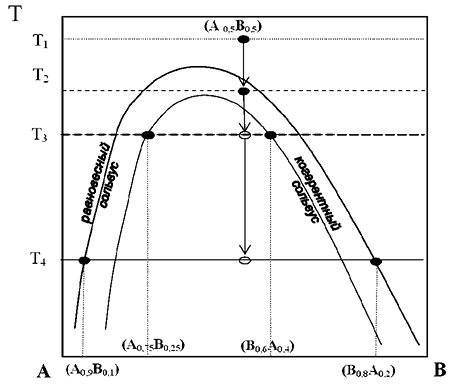
Рис. 9. Эволюция охлаждающегося твердого раствора состава (A0,5B0,5)
на субсолидусной диаграмме «состав — температура» двухкомпонентной системы A–B.
При температуре T = T1 твердый раствор стабилен. После пересечения кривой
равновесного сольвуса твердый раствор находится в метастабильном состоянии (T = T2).
Далее зародившиеся при пересечении когерентного сольвуса микровростки,
обогащенные компонентом A, достигают при T = T3 состава (A0,75B0,25)
и находятся в преобладающей по объему матрице состава (B0,6A0,4).
При дальнейшем охлаждении когерентность нарушается и при T = T3 фазы
в срастании характеризуются составами (A0,9B0,1)
и (B0,8A0,2) с небольшим преобладанием последней
Кроме распадового «сценария» поведения твердого раствора, на фоне снижения температуры возможны процессы упорядочения структуры, при которых изоморфные атомы разных элементов, имевшие в структуре твердого раствора статистическое (случайное) распределение в позициях одного типа, начинают перераспределяться в этих позициях, избегая соседства с себе подобными. Перераспределение изоморфных атомов, осуществляемое путем диффузии на малые расстояния, приводит к образованию закономерных конфигураций, так что ранее симметрически эквивалентные позиции, занимавшиеся ионами разных элементов в случайном порядке, становятся неравноценными и как бы расщепляются на два типа или более (рис. 10). Процессы упорядочения представляют собой весьма специфичные непрерывные полиморфные переходы (обычно второго рода), практически всегда приводящие к понижению симметрии и кратному увеличению элементарной ячейки (появляется сверхструктура). В некоторых случаях процессы распада и упорядочения совмещаются. Так, например, твердые растворы высокотемпературных щелочных полевых шпатов при постепенном охлаждении испытывают распад с выделением натрового компонента в виде пертитовых вростков альбита, тогда как обогащенная калием матрица, испытывая упорядочение Si и Al по тетраэдрическим позициям, постепенно превращается из моноклинного ортоклаза в триклинный микроклин.
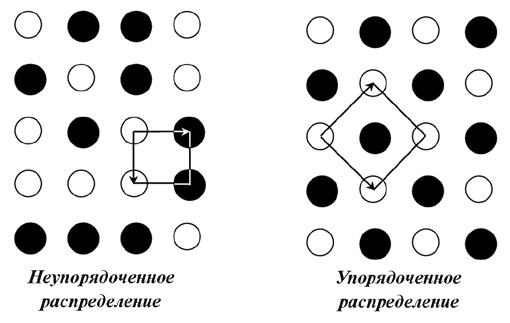
Рис. 10. Неупорядоченное (статистическое) и упорядоченное распределение
атомов двух сортов. При упорядочении элементарная ячейка изменяет
ориентацию и увеличивает площадь вдвое
Выделение новообразованных минеральных фаз из твердых растворов может начинаться с появления чрезвычайно мелкодисперсных, обычно — ориентированных включений, в результате чего образуются твердые аналоги коллоидных растворов — кристаллозоли (черный железистый сфалерит, пигментированный мельчайшими частицами пирротина). В дальнейшем новообразования испытывают тенденцию к укрупнению, они растут подобно обычным кристаллам, выпадающим из пересыщенных жидких растворов, с той лишь разницей, что транспортировка компонентов к ним осуществляется на несколько порядков медленнее, так как диффузия в твердом теле затруднена.
Процессы зарождения и укрупнения новообразованных фаз, хорошо протекающие при постепенном падении температуры (в эндогенной обстановке), могут быть приостановлены или существенно замедлены глубоким и резким охлаждением (например, при вулканизме), которое замедляет процессы диффузии и роста вновь образующихся фаз, так что твердый раствор очень долго потом сохраняется в метастабильном состоянии (например, санидины и анортоклазы из вулканических пород).
Микроиндивиды новообразованных минералов могут в ряде случаев не только не достигнуть крупных размеров, но и, напротив, уменьшиться или вовсе раствориться заново; более того, похожая участь иногда постигает и первичные включения. Подобные явления «рассасывания» механических примесей возможны, если кристалл минерала-хозяина подвергнется длительному нагреву, например, в условиях прогрессивного метаморфизма.
Следует отметить и вариант возникновения неоднородностей в минералах в процессе внешне обусловленных химических реакций, в частности как результат разложения химического соединения в связи с изменениями в окружающей среде (например, в оловосодержащем сфалерите — (Zn,Sn)S — в окислительной обстановке возникают мельчайшие включения оксида олова — SnO2). Особенно часто включения посторонних минералов как остатков от замещения (реликтов) наблюдаются в кристаллах, образующихся метасоматическим путем в метаморфических породах.
Вполне понятно, что данные химического анализа таких минералов не будут точно укладываться в химические формулы со стехиометрическими отношениями компонентов. С другой стороны, неоднородности в минералах несут с собой бесценную информацию об условиях формирования и последующих изменениях минерала, представляя собой одно из важнейших проявлений типоморфизма.
Водные соединения. Предварительно необходимо сделать следующее важное замечание. К числу водных соединений следует относить только такие, которые в своем составе содержат электрически нейтральные молекулы воды. Раньше под видом водных соединений рассматривали также минералы, содержащие гидроксильные анионы [ОН]1–. Однако между молекулой Н2О и отрицательно заряженным ионом [ОН]1– существует, естественно, принципиальная разница, весьма существенно сказывающаяся на физических и химических свойствах минералов. Гидроксил как ион способен заменять в соединениях такие анионы, как F1–, Cl1–, и прочно удерживается в кристаллических структурах. Молекула Н2О этим свойством не обладает и, будучи слабо связана в структуре, легко удаляется при нагревании. То, что гидроксил при прокаливании минералов, его содержащих, покидая кристаллическую решетку, способен превращаться в пары воды, отнюдь не может служить основанием для отождествления его с молекулой Н2O. Поэтому весьма важно в химических формулах минералов раздельно указывать присутствие в них гидроксила и воды. С этой точки зрения, например, малахит — Cu2[CO3][ОН]2 — является не водным, а основным безводным карбонатом меди, хотя при химическом анализе гидроксил определяется в виде Н2О. То же относится к минералам, представляющим собой кислые соли, в которых в числе катионов находится водород или катион Н3О+ (оксоний).
В зависимости от того, каким способом удерживается вода в минералах, различают: 1) кристаллизационную, или связанную, воду, входящую в кристаллические структуры минералов; 2) свободную воду, не участвующую в строении самого кристаллического вещества.
Связанная вода в кристаллической структуре участвует в виде молекул Н2О, занимающих в ней строго определенные места. Количество молекул воды находится в простых отношениях к другим компонентам соединения. В качестве примеров можно привести Na2CO3 . 10H2O (сода), Ca[SО4] . 2H2O (гипс), Ni3[AsO4]2 . 8H2O (аннабергит), Аl2[РО4](ОН)3 . 5Н2О (вавеллит) и др. Это так называемые кристаллогидраты, которые, по Вернеру, должны рассматриваться как «комплексные соединения», т. е. такие, в которых молекулы воды как структурные единицы располагаются в определенной координации вокруг каких-либо ионов, создавая таким путем своего рода комплексные ионы.
Так, в кристаллической структуре соединения Ni[SО4] . 6H2O рентгенометрическими исследованиями установлено, что шесть дипольных молекул Н2О непосредственно окружают катион Ni2+, ориентируясь, очевидно, определенным образом по отношению к катиону (двумя протонами Н1+ к периферии комплексного иона). Так как молекула Н2О сама по себе электрически нейтральна, то гидратированный катион [Ni(H2O)6]2+ сохраняет заряд Ni2+. Поэтому химическую формулу соединения правильнее писать так: [Ni(H2O)6][SO4].
На вопросе о причине гидратации ионов в кристаллических структурах мы остановимся позднее (во введении к разделу о кислородных солях). Здесь можно лишь указать, что необходимость гидратации ионов кристаллохимически строго оправдывается; для образования устойчивых кристаллических структур из таких крупных по размерам анионов, как [SO4]2–, присутствующие в растворе катионы Ni2+ слишком малы, в силу чего и возникает стремление к увеличению их объема без изменения заряда. Само собой разумеется, что образование кристаллогидратов может происходить лишь в средах, богатых водой, и при низких температурах.
При нагревании кристаллогидраты легко обезвоживаются, если не сразу целиком, то скачкообразно, периодически теряя часть молекул воды. При этом перестройка структуры происходит с сохранением рациональных отношений числа молекул Н2О и основного соединения. Например, халькантит Сu[SО4] . 5Н2О при искусственном обезвоживании образует вначале Cu[SО4] . 3H2O, затем Cu[SО4] . H2O и, наконец, Cu[SО4]. При этом скачкообразно меняются и такие физические свойства, как показатели преломления, удельный вес и др. Из разных соединений вода удаляется при различных температурах: некоторые из них теряют ее при комнатной температуре (многоводные сульфаты меди и железа), другие — при более высоких и даже при температурах выше 100 °С.
Свободная вода, присутствующая в минеральных массах, характеризуется тем, что не принимает прямого участия в строении кристаллического вещества минералов. При нагревании она выделяется постепенно. Различают три вида свободной воды: а) цеолитную; б) коллоидную; в) гигроскопическую.
Цеолитная вода получила свое название от общего названия особой группы минералов — цеолитов, в которых наиболее ярко проявлены особенности ее нахождения. Установлено, что молекулы воды в этой группе минералов не занимают какого-либо строго определенного положения в кристаллической структуре, а располагаются лишь в свободных полостях в ней (вдоль каналов, в межслоевых пространствах и пр.). Поэтому «растворимость» воды в них существенно ограничена. Интересно, что количество воды в них может меняться без нарушения кристаллической однородности вещества с постепенным изменением физических свойств: степени прозрачности, показателей преломления, удельного веса и др. Это указывает на то, что вода находится как бы в состоянии твердого раствора. При нагревании она выделяется в интервале температур 80–400 °С. Любопытно, что обезвоженные осторожным нагреванием цеолиты вновь способны поглощать воду с восстановлением прежних своих физических свойств.
Коллоидная вода, как показывает само название, распространена в гидрогелях, где удерживается на поверхности дисперсных фаз очень слабыми силами связи. Она по существу является адсорбированной водой, и ее наличие не зависит от структуры адсорбента (сам адсорбент, конечно, может содержать кристаллохимически связанную воду). Примером может являться опал (гидрогель кремнезема) — SiO4 . aq (aq — первые две буквы латинского слова aqua — вода). Такое обозначение коллоидной воды, принимаемое некоторыми авторами, следует признать рациональным.
Гигроскопическая (капиллярная) вода удерживается в тонких трещинах, порах и порошковатых массах силами поверхностного натяжения. В большей своей части она легко удаляется при нагревании до 100–110 °С. Резкой границы между капиллярной и коллоидной водой провести нельзя.
Кроме того, мы должны иметь в виду механические примеси воды в виде мельчайших газово-жидких включений, захваченных кристаллами во время их роста. Они широко распространены во многих минералах.
2.3. Физические свойства минералов
Уже указывалось, что минералы как физические тела обладают широким разнообразием таких свойств, как цвет, твердость, блеск, удельный вес и др. В зависимости от химического состава и кристаллической структуры эти свойства у различных минералов проявляются по-разному. Каждый минерал характеризуется какими-либо особыми признаками, по которым его можно всегда отличить от других.
Очень многие минералы можно совершенно точно определить по комплексу характерных физических свойств, не прибегая к более трудоемким исследованиям, как, например, химический анализ, рентгеноанализ и др. Нужно заметить, что для многих минералов существуют специфические, только каждому из них в отдельности свойственные тонкие особенности, которые при первом знакомстве нелегко схватить и передать словами. Особенно это относится к оттенкам цвета, густоте окраски, характеру излома, блеска и пр. Тем не менее уже при некотором опыте глаз настолько привыкает улавливать эти характерные свойства минералов, что в дальнейшем они служат решающими диагностическими признаками. Во времена далекого прошлого, когда люди еще не имели никакого представления ни о химии вообще, ни о химических элементах в частности, эти особые признаки минералов были хорошо известны и «рудознатцы» по ним безошибочно находили те полезные ископаемые, которые для них в то время представляли ценность.
Нельзя, конечно, думать, что таким путем могут быть определены все встречающиеся в природе минералы. Многие из них для окончательного установления требуют более детальных исследований, в частности применения качественных химических реакций, более точного определения удельного веса, оптических, механических и прочих свойств. Тонкозернистые минеральные массы изучаются в специальных препаратах (шлифах) под микроскопом. Очень часто в случаях установления с помощью спектрального анализа примесей таких ценных металлов, как кобальт, индий, кадмий, литий, цезий и др., имеющих промышленное значение даже в случае незначительного их содержания в минералах, приходится обращаться к химическому анализу. При изучении скрытокристаллических минеральных образований необходимо прибегать также к рентгенометрическим исследованиям. Особые методы применяются при изучении радиоактивности минералов, пьезоэлектрических эффектов, магнитных свойств и других физических явлений в минералах.
Ниже мы остановимся на разборе главнейших свойств минералов, которые имеют наибольшее диагностическое значение. К этим свойствам относятся следующие: морфологические особенности — облик кристаллов, двойники, штриховатость граней; оптические3 — прозрачность, цвет минералов, цвет черты, блеск; механические — спайность, излом, твердость, хрупкость, ковкость, упругость; а также такие свойства, как удельный вес, магнитность, радиоактивность и др.
Морфологические особенности кристаллов минералов
В природе минералы в главной своей массе распространены в виде неправильной формы зерен, не имеющих кристаллических граней, но обладающих независимо от своей формы и размеров внутренним кристаллическим строением. Хорошо образованные кристаллы, т. е. индивиды, ограниченные естественными гранями, встречаются несравнимо реже. Находки их представляют интерес в том отношении, что в распоряжении исследователя оказывается больше признаков, по которым может быть определен минерал. Существуют даже специальные определители минералов по их кристаллографическим формам.
Морфология кристаллов и учение о симметрии подробно излагаются в специальных курсах кристаллографии. Мы здесь остановимся лишь на некоторых общих особенностях морфологии кристаллов и их граней, также имеющих некоторое практическое значение при определении минералов.
Облик кристаллов. Исходя из того, что любое тело в пространстве имеет три измерения, мы среди разнообразных форм кристаллов и кристаллических зерен прежде всего должны выделить следующие основные типы.
1. Изометрические формы, т. е. формы, одинаково развитые во всех трех направлениях в пространстве. Примером их могут служить ромбододекаэдры граната, октаэдры магнетита, кубы пирита (рис. 11) и др. Изометрический облик особенно характерен для минералов кубической сингонии, но и кристаллы минералов других сингоний также могут обладать обликом, приближающимся к изометрическому (рис. 12).
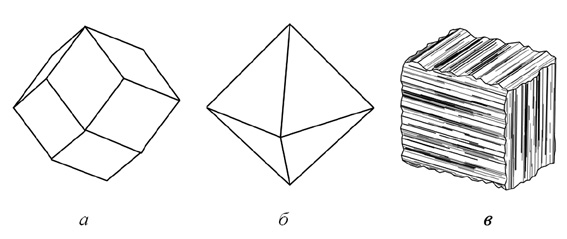
Рис. 11. Кристаллы изометрического облика минералов кубической сингонии:
а — ромбододекаэдр граната; б — октаэдр магнетита
и в — куб пирита с комбинационной штриховкой
Рис. 12. Кристаллы изометрического облика минералов тетрагональной (а и б)
и тригональной (в) сингонии: а — шеелит; б — анатаз и в — шабазит
2. Формы, вытянутые в одном направлении, т. е. призматические, столбчатые, шестоватые, игольчатые (рис. 13), волосистые кристаллы, волокнистые образования. Например, кристаллы аквамарина, турмалина и др.
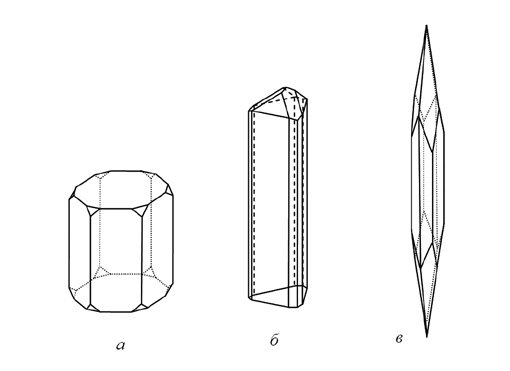
Рис. 13. Кристаллы: а — столбчатого (берилл); б — шестоватого (турмалин)
и в — игольчатого (антимонит) облика
3. Формы, вытянутые в двух направлениях при сохранении третьего короткого. Сюда следует отнести таблитчатые, пластинчатые, листоватые и чешуйчатые кристаллы Таковы, например, наблюдающиеся кристаллы гематита (Fe2O3), слюд (рис. 14) и др.
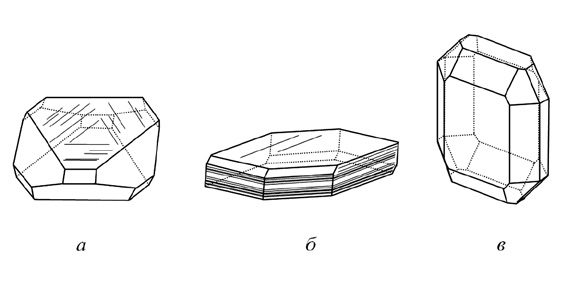
Рис. 14. Кристаллы: а — уплощенного (гематит);
б — пластинчатого (слюда) и в — таблитчатого (оливин) облика
Широко распространены и переходные между этими основными типами формы. Таковы, например, досковидные кристаллы кианита (Al2SiO5), имеющие промежуточную форму между вторым и третьим типами (уплощенные столбчатые кристаллы); бочонковидные кристаллы корунда (Al2О3) или скаленоэдрические кристаллы кальцита (Са[СО3]) как промежуточные формы между первым и вторым типами; формы, приближающиеся к линзовидным (промежуточным между первым и третьим типами), — уплощенные кристаллы титанита (CaTi[SiO4]O), монацита (Се[РО4]) и др.
Кроме того, существуют сложные и искаженные формы кристаллов, например блочные (рис. 15), расщепленные (рис. 16) и скрученные кристаллы (рис. 17), сферокристаллы (рис. 18) и нитевидные кристаллы (усы, антолиты) (рис. 19).
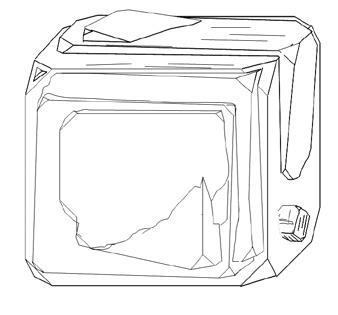
Рис. 15. Блочный кристалл пирита
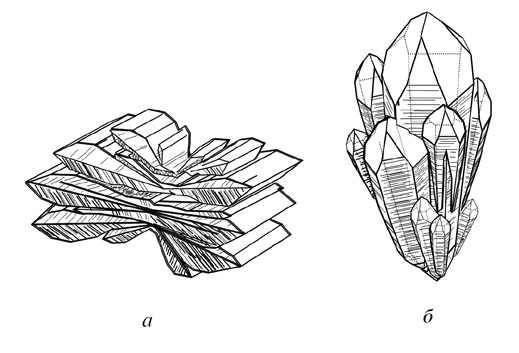
Рис. 16. Плоскорасщепленный кристалл барита (а)
и объемнорасщепленный кристалл кварца (б)

Рис. 17. Скрученный мозаичноблочный кристалл пирита.
Рисунок В. Слетова и В. Макаренко из III выпуска альбома «Рисуя минералы... » (рис. 21)
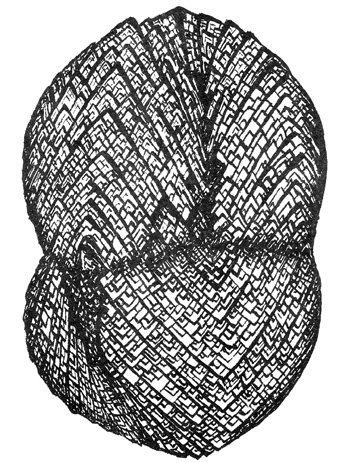
Рис. 18. Сферокристалл сидерита. Рисунок В. Слетова
из II выпуска альбома «Рисуя минералы...» (рис. 12)
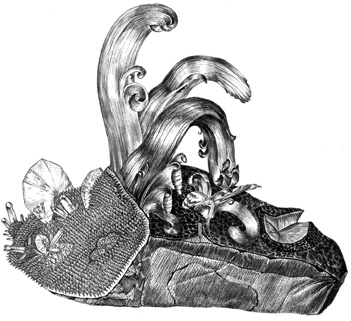
Рис. 19. Гипсовые антолиты — одна из разновидностей нитевидных кристаллов;
образуются при кристаллизации на пористой подложке.
Рисунок В. Слетова и В. Макаренко из I выпуска альбома «Рисуя минералы...» (рис. 11)
Преимущественное развитие отдельных элементов огранения кристаллов приводит к отклонениям от выпуклой формы, при этом образуются скелетные кристаллы (реберные формы (рис. 20) и вершинные ветвящиеся формы — кристаллические дендриты (рис. 21)). Известны также кристаллы с антискелетным типом развития (рис. 22).
Рис. 20. Реберные скелетные кристаллы: а — нашатырь; б — галит
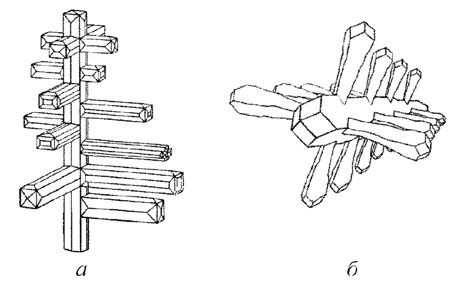
Рис. 21. Вершинные скелетные кристаллы: а — медь; б — нашатырь
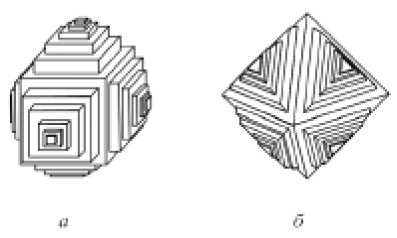
Рис. 22. Антискелетные кристаллы: а — флюорит; б — магнетит
Помимо облика кристаллических индивидов различают также габитус кристаллов, относящийся лишь к хорошо ограненным минералам4. Характеристика габитуса основывается на преобладании тех или иных кристаллографических форм в кристаллах данного минерала. Например, кристаллы галенита (PbS) обычно встречаются в виде кубов, у которых иногда углы слегка притуплены гранями октаэдра, реже — кубооктаэдров и изредка — октаэдров, слегка притупленных гранями куба (рис. 23). Общая форма (облик) для всех них является изометрической, однако габитус кристаллов различен: у первого преобладают или исключительно развиты грани куба, у третьего, наоборот, преимущественно развиты грани октаэдра, а у второго — те и другие образованы примерно в одинаковой степени. По-видимому, на образовании того или иного габитуса кристаллов сказывается влияние особенностей состава среды, в которой происходит минералообразование.
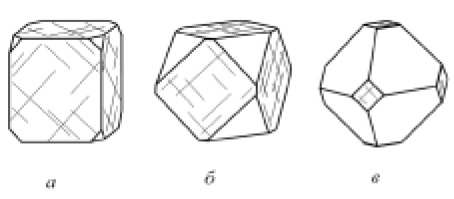
Рис. 23. Кристаллы галенита различного габитуса: а — кубический габитус
с подчиненным развитием октаэдра; б — кубооктаэдрический габитус;
в — октаэдрический габитус с подчиненным кубом
Хотя далеко не все минералы сразу легко узнать по формам их кристаллов, для ряда минералов форма кристаллов настолько характерна, что она является важнейшим диагностическим признаком. Например, призматические кристаллы кварца, усеченные гранями ромбоэдра и трапецоэдра, всегда легко узнаются независимо от того, в какой цвет они окрашены. Типичны также кубические или пентагон-додекаэдрические кристаллы пирита, октаэдрические кристаллы шпинели, магнетита, ромбододекаэдрические кристаллы граната и др.
Характерные черты форм кристаллов нашли свое отражение в самих названиях ряда минералов. Примеры: актинолит (по-гречески — лучистый камень), гранат (от лат. granum — зерно), лепидолит (от греч. лепис — чешуя), аксинит (от греч. аксине — топор), корундофиллит (от греч. филлон — лист), хризотил (по-гречески — золотистое волокно) и др.
Приведем названия и международные обозначения 32 видов симметрии кристаллов (табл. 5).
Таблица 5. Названия и международные символы
32 видов симметрии (точечных групп) кристаллов
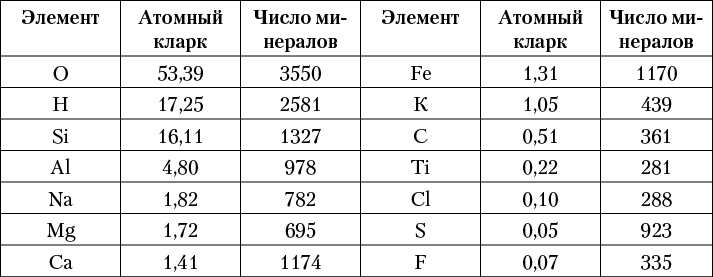
Двойники и закономерные сростки. Двойником, как известно, называют закономерный сросток двух кристаллов одного и того же минерала, в котором индивиды могут быть совмещены друг с другом либо поворотом вокруг некоторой оси, не принадлежащей к числу осей симметрии данного кристалла (рис. 24а), либо отражением в плоскости симметрии (рис. 24б), либо путем инверсии. В случаях закономерного срастания трех индивидов сростки носят название тройников, четырех индивидов — четверников и т. д. Как показал Н. В. Белов, двойниковая плоскость очень часто совпадает с плоскостью плотнейшей упаковки ионов. Таков, например, шпинелевый закон срастания кристаллов многих минералов кубической сингонии по (111).
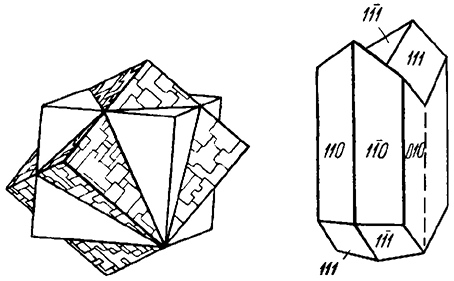
Рис. 24. Двойники: а — по оси третьего порядка (флюорит); б — по плоскости (100) (гипс)
В большинстве случаев двойники можно распознать по такому их характерному признаку, как входящие углы на поверхности кристаллов (характерные также и для грубоблочных кристаллов). Однако в ряде случаев приходится прибегать к искусственному травлению, а наиболее надежные результаты доставляют кристаллооптические исследования в поляризованном свете.
Образование двойников может происходить: 1) путем срастания зародившихся кристалликов в растворе при их соприкосновении во время роста; 2) в связи с механическими воздействиями (при одностороннем внешнем давлении); 3) при полиморфных превращениях кристаллического вещества; 4) при нарушении порядка следования слоев в плотноупакованных структурах (ростовые ошибки упаковки).
Для некоторых минералов двойниковые образования являются типичными и нередко облегчают их диагностику. Таковы, например, коленчатые двойники рутила (TiO2) и касситерита (SnO2), так называемые «ласточкины хвосты» гипса (см. рис. 24б), тройники хризоберилла (ВеАl2О4), крестообразные двойники ставролита (от греч. ставрос — крест) и др.
Закономерные сростки различных минералов также давно описывались в литературе. Закономерная ориентировка срастающихся минералов обусловлена общностью или близостью строения плотно упакованных плоскостей срастания. Наблюдаются разные случаи таких сростков: 1) эпитаксия — образовавшиеся кристаллы одного минерала обрастаются другим (например, кристаллы блеклой руды — Cu3SbS3 — покрываются закономерно ориентированными кристалликами или сплошной «рубашкой» халькопирита — CuFeS2); 2) эндотаксия — закономерно ориентированные вростки, например, ильменита (FeTiO3) в кристаллических зернах магнетита (FeFe2O4) как продукт распада твердого раствора (устанавливаются в полированных шлифах под микроскопом); 3) гомоосевой псевдоморфизм — ориентированное замещение с периферии одного минерала другим (например, сфалерита — ZnS халькопиритом — CuFeS2 с сохранением даже двойникового строения замещаемого минерала) и др.
Скульптура граней кристаллов. Как мы знаем, грани кристаллов, обозначаемые простыми символами, не представляют собой идеальных плоскостей. При рассматривании их в отраженном свете (особенно при увеличении) почти всегда можно обнаружить те или иные дефекты: неровности поверхности, вицинали, штриховатость, фигуры травления и пр., обусловленные, по-видимому, неравномерной скоростью роста кристаллов или их частичным растворением в связи с изменением концентрации компонентов в остаточном растворе, колебаниями температуры, иногда механическими нарушениями в кристаллах и др.
Штриховатость для ряда минералов — широко распространенное явление, которое может служить важным диагностическим признаком. У одних минералов она проявляется вдоль вытянутости кристаллов, например у турмалина, эпидота, у других — поперек, например на призматических гранях кварца. Для кубических кристаллов пирита (см. рис. 11в) весьма характерно, что штрихи одной грани расположены перпендикулярно по отношению к каждой соседней грани.
Штриховатость граней может быть различного происхождения: 1) комбинационная, обусловленная многократным повторением узких граней (алмаз, турмалин); 2) двойниковая — как результат полисинтетического сложения кристаллов (сфалерит, иногда плагиоклазы и др.); 3) индукционная, обусловленная взаимным влиянием соприкасающихся одновременно растущих кристаллов.
Фигуры травления на гранях кристаллов являются результатом начальной стадии растворения кристаллов. Согласно экспериментальным исследованиям вершины и ребра кристаллов растворяются быстрее, чем грани. Как показал И. И. Шафрановский, в природных кристаллах в результате частичного растворения нередко образуются конусовидные образования на кристаллах ряда минералов (кварца, топаза (рис. 25) и др.). Им же на кристаллах алмаза было установлено наличие идеально правильных конусов вокруг четверных и тройных осей симметрии с образованием округлых ромбододекаэдров («додекаэдроидов»).
Рис. 25. Конусы растворения на кристалле топаза
Фигуры травления на гладких гранях кристаллов разных минералов обладают различной симметрией, что связано с их кристаллической структурой. Поэтому нередко по ориентировке фигур травления, например на кристаллах кварца, можно доказать их принадлежность к правому или левому кварцу, иногда наличие двойникового строения и пр.
Прозрачностью называется свойство вещества пропускать сквозь себя свет. Абсолютно непрозрачных тел не существует, однако многие минералы, особенно металлы (даже в тонких пленках), видимые лучи пропускают в столь малых количествах, что практически кажутся совершенно непрозрачными. Точно так же не существует и абсолютно прозрачных материальных сред, т. е. таких, которые совершенно не поглощали бы пропускаемого через них света. Одна из самых прозрачных сред — чистая вода — в толстом слое имеет явно голубой цвет, что свидетельствует о существенном поглощении лучей красного конца спектра видимого света.
Из курса физики мы знаем о том, что одна часть падающего на данное тело светового потока отбрасывается или отражается, а другая вступает внутрь среды. Оставим пока в стороне явления отражения света (к ним мы вернемся в разделе о блеске минералов), а здесь рассмотрим поведение луча, вступившего в среду.
Как известно, вступивший в данную среду луч света меняет свою скорость, преломляется и по мере проникновения вглубь постепенно расходует свою энергию на превращение ее в другие виды энергии (преимущественно тепловую), благодаря чему количество света постепенно уменьшается, т. е. происходит поглощение (абсорбция) света.
Таким образом, интенсивность вышедшего из данной среды света I будет более ослабленной по сравнению с интенсивностью вступившего света I0.. Иначе говоря, отношение I : I0 = a будет всегда правильной дробью. Величина a называется коэффициентом прозрачности данной среды при толщине слоя, равной единице (1 см). Она зависит от химической природы вещества и длины волны света (но не от силы света). Чем ближе эта величина к единице, тем более прозрачен минерал, и наоборот.
В зависимости от степени прозрачности все минералы, наблюдающиеся в крупных кристаллах, делят на следующие группы:
1) прозрачные — горный хрусталь, исландский шпат, топаз и др.;
2) полупрозрачные — изумруд, сфалерит, киноварь и др.;
3) непрозрачные — пирит, магнетит, графит и др.
Многие минералы, кажущиеся в больших кристаллах или обломках непрозрачными, просвечивают в тонких осколках или тонких шлифах (биотит — черная слюда, рутил и др.).
Когда мы вместо крупных кристаллов имеем дело с тонкозернистыми агрегатами, наблюдается иная картина в отношении прозрачности веществ. Если тело состоит из множества маленьких частиц — зерен, различно оптически ориентированных, то в такой среде лучи света не могут проложить себе прямых длинных путей. Свет в подобных средах, многократно преломляясь в различных направлениях, в конце концов рассеивается и отражается. Поэтому такие среды кажутся непрозрачными. В этом легко убедиться, если сравнить пластинку прозрачного кальцита (исландского шпата) и такой же толщины отполированную с обеих сторон пластинку тонкозернистого белого мрамора, состоящего из агрегата кальцитовых зерен. В то время как сквозь исландский шпат мы легко можем читать надписи на этикетке, пластинка мрамора не пропускает света. Только в тонких шлифах такие тела обнаруживают свою прозрачность.
Если при этом вещество, обладающее тонкоагрегатным строением, не проявляет заметного поглощения света, то для него характерен молочно-белый цвет. Наиболее резко этот цвет выражен в тех случаях, когда вещество находится в дезагрегированном состоянии, т. е. когда в промежутках между мельчайшими обломками или частицами присутствует воздух. Это явление нам хорошо знакомо: если мы ударим молотком по прозрачному голубоватому льду, то в местах удара появляется молочно-белая окраска благодаря возникновению массы тончайших трещинок и пустот, выполненных воздухом.
Окраска минералов невольно обращает на себя внимание при первом же знакомстве с ними и потому является одним из важнейших признаков, свойственных минералам.
Вполне естественно поэтому, что многие названия даны минералам именно по этому признаку. Примеры: лазурит, азурит (от франц. azur — лазурь), хлорит (от греч. хлорос — зеленый), родонит (от греч. родон — розовый), рубин (от лат. ruber — красный), крокоит (от греч. крокос — шафран, т. е. здесь имеется в виду его красно-оранжевый цвет), аурипигмент (от лат. aurum — золото), хризолит, хризоберилл (от греч. хризос — золото), эритрин (от греч. эритрос — красный), гематит (от греч. гематикос — кровавый), альбит (от лат. albus — белый), меланит (от греч. мелас — черный) и т. д. И наоборот, такие названия, как «киноварь», «малахитовая зелень» и др., вошли в наш язык как стандартные цвета красок, что говорит о том, что эти цвета постоянно присущи данным минералам.
В целом проблема окраски минералов очень сложна. Хотя наши познания в области причин появления окрасок кристаллических веществ благодаря большим успехам физики и кристаллохимии в последнее время значительно подвинулись вперед, все же остается еще много неясных вопросов. Первую более обстоятельную попытку обобщить имеющийся материал по этому вопросу и увязать окраску природных соединений с их кристаллохимическими особенностями сделал А. Е. Ферсман в своей книге «Цвета минералов» (1937). Дальнейшие достижения в исследовании причин окраски и механизмов формирования цвета минералов отражены в книге А. Н. Платонова «Природа окраски минералов» (Киев, Наукова думка, 1976).
В природных химических соединениях различают три рода окрасок по происхождению: 1) идиохроматическую; 2) аллохроматическую; 3) псевдохроматическую.
Идиохроматизм5. Во многих случаях окраска природных соединений, никогда не встречающихся в виде бесцветных кристаллов, обусловлена внутренними свойствами самого минерала (его конституцией). Таковы, например, черный магнетит (FeFe2O4), латунно-желтый пирит (FeS2), карминно-красная киноварь (HgS), зеленые и синие кислородные соли меди (малахит, азурит, бирюза и др.), густо-синий лазурит и т. д.
Эти типичные окраски минералов получили название идиохроматических. В различных минералах они обусловлены разными причинами.
1. В многочисленных минералах окраска обязана своим происхождением тому, что в состав самих соединений входит какой-либо хромофор, т. е. химический элемент, приносящий окраску. К числу их, как давно уже было установлено, относятся следующие: Ti, V, Cr, Mn, Fe, Co, Ni, т. е. элементы семейства железа, располагающиеся в центре менделеевской таблицы элементов, разбитой по длинным периодам, и в меньшей степени — W, Mo, U, Сu и TR.
Наиболее ярким представителем хромофоров является хром, само название которого указывает на эту его особенность6. Содержание хрома в минералах обусловливает весьма интенсивные окраски — красную (пироп, рубин и др.), ярко-зеленую (уваровит, изумруд, фуксит), фиолетовую (родохром, кеммерерит). Насколько сильным красителем является хром, можно судить по тому, как изоморфная примесь окиси хрома в количестве всего лишь 0,1 % окрашивает бесцветное соединение — окись алюминия — в густой ярко-красный цвет. При этом содержании расстояние между двумя ближайшими частицами хромофоров в массе корунда составит около 20 Å , т. е. во много раз больше, чем радиусы самих ионов (0,57 Å у Аl и 0,64 Å у Сr). Очевидно, ионы хрома, сильно отличаясь от ионов алюминия по конфигурации электронных оболочек, создают вокруг себя сильные нарушения в симметрии электрического поля и, несмотря на состояние рассеяния, оказывают свое влияние на всю структуру соединения.
Указанные выше зеленые и фиолетовые окраски минералов обусловлены значительным содержанием Cr2O3 (до нескольких процентов и даже десятков процентов). Сама чистая окись хрома также окрашена в зеленый цвет. Однако нельзя утверждать, что причиной зеленой окраски всегда являются большие содержания хрома в химических соединениях. Установлено, например, что зеленые бериллы — изумруды — обязаны своей чудесной яркой окраской совершенно ничтожной изоморфной примеси Сr2О3 к Аl2О3 в пределах нескольких сотых процента, а нередко зеленая окраска изумрудов связана отчасти и с присутствием V2O3. Следовательно, явление окраски минералов далеко не столь простое, как это могло бы показаться с первого взгляда.
Ион [CrO4]2–, содержащий Сr6+, в искусственных соединениях обычно дает желтые соединения, но в соединении его с сильно поляризующими катионами наблюдается окрашивание в густой оранжево-красный цвет. Таков, например, минерал крокоит (РЬ[CrO4]).
2. В некоторых случаях минерал может быть окрашен в тот или иной цвет вне всякой связи с хромофорами или с изменением его химического состава, так как химическим и спектральным анализами не удается установить хотя бы ничтожные следы каких-либо примесей красящего пигмента.
Так, известны случаи окраски каменной соли (NaCl) в красивый синий цвет. Оказалось, что эта окраска своим происхождением обязана тому, что часть ионов этого соединения, в частности натрия, превратилась в нейтральные атомы, присоединив к себе необходимые для этого электроны. Это один из случаев так называемой дефектной окраски (активными центрами, вызывающими специфическое поглощение света в некоторых участках спектра, являются дефекты структуры). Такую окраску в прозрачной каменной соли легко вызвать искусственно. Для этого достаточно пропитать ее парами металлического натрия. Точно такой же результат можно получить, если воздействовать на каменную соль катодными лучами, приносящими с собой свободные электроны.
Следовательно, окраска некоторых прозрачных минералов может быть связана с изменением однородности строения кристаллических структур, с изменением электростатического состояния ионов, могущих превращаться под влиянием тех или иных причин в нейтральные атомы или в возбужденные (слабо заряженные) атомы. В сущности, окраска минералов такого происхождения, обусловленная привходящими причинами, не имеющими прямого отношения к конституции минерала, т. е. не присущая всем его индивидам без исключения, должна быть отнесена к аллохроматической (см. ниже).
3. Особую, хотя и очень небольшую, группу окрашенных минералов составляют такие соединения, в которых окраска обусловлена не наличием хромофоров и не нарушением электростатической однородности кристаллических структур, а присутствием ионов или целых групп их внутри пустых промежутков структуры. Это относится, в частности, к тем силикатам, у которых имеет место «внедрение» таких дополнительных анионов, как Cl1–, [SO4]2– и др. Примером здесь может служить ярко-синий минерал лазурит.
Истинная природа этого явления, получившего название стереохроматизма, еще не разгадана, но ясно то, что окраска таких минералов, несомненно, связана с группами присоединения или внедрения, которые сами по себе не являются хромофорами. Характерны прочность и стойкость в огне этих окрасок, которые были известны еще за несколько тысячелетий до нашей эры, когда лазурит применялся в качестве краски.
Аллохроматизм7. Известно немало примеров, когда один и тот же минерал бывает окрашен в различные цвета и оттенки. Так, кварц, обычно встречающийся в виде бесцветных, часто совершенно прозрачных кристаллов (горный хрусталь), бывает окрашен в красивый фиолетовый цвет (аметист), розовый, желто-бурый (от окислов железа), золотистый (цитрин), серый или дымчатый (раухтопаз), густой черный (морион), наконец, в молочно-белый цвет. Точно так же каменная соль — галит — может обладать белым, серым, желтым, бурым, розовым и иногда синим цветом.
В большинстве случаев окраска в таких минералах связана с посторонними тонкорассеянными механическими примесями, окрашенными в тот или иной цвет хромофорами (носителями окраски). Эти красящие вещества могут быть представлены как неорганическими, так и органическими соединениями. Часто бывает достаточно совершенно ничтожного их количества для того, чтобы вызвать интенсивное окрашивание бесцветных минералов, причем окраска зависит не только от количества, но и от степени дисперсности этих веществ.
Подобные окраски, не зависящие от химической природы самого минерала, носят название аллохроматических (т. е. чуждых самим минералам). Окрашенные таким способом минералы представляют собой не что иное, как кристаллозоли.
Наблюдаются и более грубые дисперсии, т. е. на глаз заметные рассеянные включения тех или иных минералов. Таковы, например, зеленоватые кристаллы кварца с микроскопическими включениями актинолита, черный кальцит, переполненный мельчайшими включениями сульфидов или углеродистого вещества, и др.
Наконец, следует указать на многие аллохроматически окрашенные полупрозрачные и непрозрачные коллоиды (гели) и метаколлоиды. В большинстве случаев к числу загрязняющих красящих примесей принадлежат бурые гидроокислы железа, красная окись железа, черные окислы марганца, органические вещества и др. Очень часто красящий пигмент в них распределен неравномерно, иногда концентрическими слоями. Таковы, например, агаты с их замечательно тонкими и разноцветными рисунками.
Псевдохроматизм8. В некоторых прозрачных минералах иногда наблюдается «игра цветов», обусловленная интерференцией падающего света в связи с отражением его от внутренних поверхностей трещин спайности, иногда от поверхности каких-либо включений. Это явление нам хорошо знакомо по иризирующим пленкам керосина, нефти, масла, плывущим по воде и окрашенным в различные «цвета радуги». Оно объясняется тем, что здесь интерферируют лучи света, отраженные от передней поверхности прозрачной пленки (ее границы с воздухом) и задней (поверхности раздела с водой).
Подобные явления ложной окраски наблюдаются и в твердых прозрачных минералах. Прекрасным примером может служить поделочный камень, относящийся к группе плагиоклазов из семейства полевых шпатов, — лабрадор, в котором, особенно на полированных плоскостях, при некоторых углах поворота вспыхивают местами красивые синие и зеленые переливы, обусловленные интерференцией света на параллельно расположенных тончайших пластинах (ламелях) различного по основности плагиоклаза. Такая микроструктура образуется при распаде первоначально гомогенного твердого раствора.
Иризирующие пестроокрашенные пленки нередко наблюдаются на почковидной поверхности бурых железняков (гидроокислов железа), кристаллах железного блеска, на слегка окислившейся поверхности борнита — Cu5FeS4 (в виде фиолетовых и синих отливов) и др. Во всех этих случаях окраска ничего общего не имеет с природой самого минерала. Подобные пленки на минералах называются побежалостями.
О классификации цветов. Необычайное разнообразие оттенков в окраске минералов не поддается сравнительному описанию не только потому, что их очень много, но и потому, что наш орган зрения непосредственно воспринимает лишь грубые различия в цветах минералов. В этом нетрудно убедиться, если сравнить между собой два таких зеленых минерала, как малахит и гарниерит (никелевый гидросиликат). При рассматривании порознь они для неопытного глаза кажутся одинаково окрашенными, но когда мы положим их рядом, то легко заметим существенное различие в оттенках окраски. В процессе постоянной тренировки в восприятии цветовых эффектов постепенно вырабатывается зрительная память на цвета, все больше и больше начинают улавливаться характерные особенности оттенков. Способность запоминать эти особенности оказывает большую услугу в диагностике минералов.
В обычной практике при определении цвета минералов прибегают к сравнительной оценке, сопоставляя его с окраской каких-либо хорошо известных предметов или веществ. Поэтому широко пользуются двойными названиями цветов минералов, например: молочно-белый, медово-желтый, латунно-желтый, карминно-красный, изумрудно-зеленый, яблочно-зеленый (цвет неспелого яблока), шоколадно-бурый, свинцово-серый, оловянно-белый и т. д. Несмотря на то что все эти определения весьма относительны, они все же приняты и встречаются во всей мировой литературе по минералогии.
Как бы то ни было, на первых порах мы должны условиться о названиях хотя бы основных цветов, прикрепив их к определенным минералам. За основу можно принять следующие часто употребляемые названия цветов, более или менее постоянные для ряда минералов:
1) фиолетовый — аметист;
2) синий — азурит;
3) зеленый — малахит;
4) желтый — аурипигмент;
5) оранжевый — крокоит;
6) красный — киноварь (в порошке);
7) бурый — пористые разности лимонита;
8) желто-бурый — охристые разности лимонита;
9) оловянно-белый — арсенопирит;
10) свинцово-серый — молибденит;
11) стально-серый — блеклая руда;
12) железно-черный — магнетит;
13) индигово-синий — ковеллин;
14) медно-красный — самородная медь;
15) латунно-желтый — халькопирит;
16) металлически-золотистый — золото.
В качестве примера ахроматических цветов, возникающих при равномерном поглощении всего спектра видимого света, приведем следующие: бесцветный горный хрусталь, молочно-белый кварц, серая каменная соль и черный пиролюзит.
Под этим термином подразумевается цвет тонкого порошка минерала. Этот порошок легко получить, если мы будем проводить испытуемым минералом черту на матовой (неглазурованной) поверхности фарфоровой пластинки, называемой бисквитом. Порошок получается в виде следа на пластинке, окрашенного в тот или иной характерный для данного минерала цвет.
Этот признак в сравнении с окраской минералов является гораздо более постоянным, а следовательно, и более надежным диагностическим признаком.
Цвет черты, или порошка, в ряде случаев совпадает с цветом самого минерала. Например, у киновари окраска и цвет порошка красные, у магнетита — черные, у лазурита — синие и т. д. Для других минералов наблюдается довольно резкое различие между цветом минерала и цветом черты. Из числа известных в природе минералов такое различие мы наблюдаем, например, у гематита (цвет минерала стально-серый или черный, черта — красная), у пирита (цвет минерала латунно-желтый, черта — черная) и т. д.
Большинство прозрачных или полупрозрачных окрашенных минералов обладают бесцветной (белой) или слабоокрашенной чертой. Поэтому наибольшее диагностическое значение цвет черты имеет для непрозрачных или полупрозрачных резко окрашенных соединений.
В природе нередко один и тот же минерал встречается то в плотных, то в порошковатых разностях. Цвета их в ряде случаев сильно отличаются друг от друга. Примерами могут служить: лимонит (гидроокись железа) — в плотных массах черный, а в порошковатых (в виде охры) — желто-бурый; гематит (безродная окись железа) — в кристаллической разновидности почти черный, а в порошковатых разностях — ярко-красный и т. д. В других случаях цвет минерала в плотных кристаллических массах и в диспергированном состоянии одинаков; например, у малахита он и в том и в другом виде зеленый, у азурита — синий, у киновари — красный, у аурипигмента — ярко-желтый и т. д.
Следует упомянуть, что аллохроматическая окраска многих полупрозрачных минералов, вызванная примесями в виде дисперсной фазы тех или иных соединений, в сущности отвечает цвету этих соединений в порошковатом состоянии. Таковы, например, желто-бурые и бурые опалы, окрашенные гидроокислами железа, красные яшмы, густо проникнутые тонкораспыленной безводной окисью железа, и т. д.
Блеск и показатель преломления
Падающий на минерал световой поток частью отбрасывается назад, причем частота колебаний не претерпевает изменений. Этот отраженный свет и создает впечатление блеска минерала. Интенсивность блеска, т. е. количество отраженного света, тем больше, чем резче разница между скоростями света при переходе его в кристаллическую среду, т. е. чем больше показатель преломления минерала. Блеск почти не зависит от окраски минералов.
Зная показатели преломления минералов, для подавляющего большинства минералов нетрудно вычислить показатель отражения света R по формуле Френеля:
 ,
,
где R — показатель отражения; N — средний показатель преломления минерала по отношению к воздуху.
Подставляя в эту формулу ряд определенных значений N, легко изобразить графическим путем — в виде кривой — зависимость показателя отражения (блеска) от показателя преломления (рис. 26). Кривая, как видим, имеет минимум для N = 1, к которому близок показатель преломления воздуха. Так как подавляющая масса минералов обладает показателями преломления выше единицы, то интересующие нас значения показателя отражения R будут располагаться вправо от этого минимума.
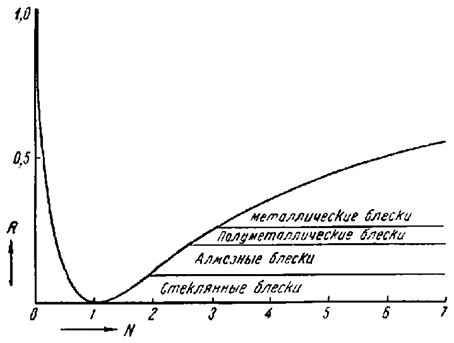
Рис. 26. Зависимость показателя отражения (R) от показателя преломления (N) минералов
Давно установленные чисто практическим путем градации интенсивностей блеска минералов почти точно укладываются в следующую ступенчатую шкалу.
1. Стеклянный блеск, свойственный минералам с N = 1,3–1,85. К ним относятся: лед (N = 1,309), криолит (N = 1,34–1,36), флюорит (N = 1,43), кварц (N = 1,544); далее следуют многочисленные галоидные соединения, карбонаты, сульфаты, силикаты и другие кислородные соли; заканчивается этот ряд такими минералами, как шпинель (N = 1,73), корунд (N = 1,77) и большинство гранатов (N до 1,85).
2. Алмазный блеск, характерный для минералов с N = 1,85–2,6. В качестве примеров сюда следует отнести: англезит (N = 1,87–1,89), циркон (N = 1,92–1,96), касситерит (N = 1,99–2,09), самородную серу с алмазным блеском на плоскостях граней (N = 2,04), сфалерит (N = 2,3–2,4), алмаз (N = 2,40–2,46), рутил (N = 2,62), часто обладающий полуметаллическим блеском, свойственным густоокрашенным разностям.
3. Полуметаллический блеск прозрачных и полупрозрачных минералов с показателями преломления (для Li-света) N = 2,6–3,0. Примеры: алабандин (N = 2,70), куприт (N = 2,85), киноварь (N = 2,91).
4. Металлический блеск минералов с показателями преломления выше 3. В порядке возрастающей отражательной способности приведем следующие примеры: гематит, пиролюзит (кристаллический), молибденит, антимонит, галенит, халькопирит, пирит, висмут и др.
Влево от минимума (см. рис. 26) кривая отражательной способности круто поднимается вверх. В эту область с показателями преломления менее единицы попадают лишь некоторые чистые (самородные) металлы: серебро (N = 0,18), золото (N = 0,36), медь (N = 0,64) и др.
Необходимо указать, что при определении отражательной способности непрозрачных минералов помимо показателей преломления нельзя не учитывать также коэффициента поглощения (K) данной среды. Для этих случаев показатель отражения (R) выражается следующей формулой (для оптически изотропных сечений минералов):
 .
.
Это означает, что для непрозрачных минералов величины показателей отражения в действительности будут несколько выше, чем это определяется по формуле Френеля. Этим легко объясняются кажущиеся редкие исключения из приведенного выше положения. Например, магнетит обладает показателем преломления 2,42, т. е. должен был бы иметь алмазный блеск, однако благодаря непрозрачности, т. е. значительному поглощению света, показатель отражения несколько повысится, перейдя на диаграмме (см. рис. 26) в полосу полуметаллических блесков.
Если мы зададимся вопросом, какие же блески в минеральном царстве преобладают, то, распределив все прозрачные и просвечивающие минералы по среднему показателю преломления (рис. 27), увидим отчетливо выраженный широкий максимум для значений 1,5–1,7. Подсчет показывает, что на долю минералов со стеклянным блеском приходится около 70 % природных соединений с показателями преломления, не превышающими 1,9. Другая группа, правда, менее многочисленная, приходится на минералы с металлическим блеском. Однако эти металлические блески настолько характерны для целого ряда важных в практическом отношении минералов, что многие из последних раньше носили название (а в немецком языке и до сих пор называются) по этому признаку; например: галенит (свинцовый блеск), халькозин (медный блеск), антимонит (сурьмяный блеск), кобальтин (кобальтовый блеск), гематит (железный блеск) и т. д.
Рис. 27. Относительная распространенность минералов
с различными показателями преломления
Показатель преломления, как известно, в общем находится в зависимости от рефракции ионов, химического состава минералов, их удельного веса и от особенностей кристаллической структуры. Давно уже было подмечено, что в минералах, обладающих одинаковой кристаллической структурой, показатель преломления, как и удельный вес, обычно увеличивается с увеличением атомного веса катиона. Например, для MgO (уд. в. 3,64) N = 1,73, а для NiO (уд. в. 6,4) N = 2,23; или для Аl2О3 (уд. в. 4,0) N = 1,76, a для Fe2O3 (уд. в. 5,2) N = 3,01 и т. д. Известно также, что вхождение в состав соединений в виде изоморфных примесей высоковалентных ионов — Fe3+, Cr3+, Ti4+, V5+ и др. — значительно повышает показатель преломления. В изоструктурных соединениях, например NaCl и КСl, увеличение размеров катиона (Na1+ 0,98 и К1+ 1,33) приводит к менее плотной упаковке размеров и даже к понижению N (для КСl —1,490, тогда как для NaCl — 1,544) в соответствии с понижением удельного веса, несмотря на то что атомный вес К (39,0) выше, чем Na (23,0). Обратная картина для показателя преломления устанавливается в соединениях NaF и NaCl, где анион фтора (ат. вес 19,0) заменяется анионом хлора (ат. вес 35,5): для NaF N = 1,328, а для NaCl N = 1,544, т. е. у первого соединения значительно ниже, чем у второго, хотя удельный вес NaF (2,79) выше, чем NaCl (2,16). Объясняется это очень низкой рефракцией фтора. Само собой разумеется также, что изменение координационного числа катиона при перестройке кристаллической структуры сказывается, как это указывалось выше (см. полиморфизм), на удельном весе, а следовательно, и на показателе преломления кристаллического вещества.
Вторым важным фактором (независимо от показателей преломления и поглощения света), влияющим на результат отражения света, является характер поверхности, от которой происходит отражение.
Выше мы рассмотрели блески минералов, обусловленные зеркально гладкими поверхностями (т. е. гранями кристаллов и плоскостями спайности). Но если минерал в изломе имеет не идеально гладкую, а скрытобугорчатую или ямчатую поверхность, то стеклянные, алмазные и другие блески приобретают чуть тусклый оттенок. Отраженный свет при этом частично теряет свою упорядоченность, подвергаясь некоторому рассеиванию. Создается жировой, или, как чаще говорят, жирный блеск. В этом явлении мы можем наглядно убедиться, если проследим за изменением блеска в свежем изломе каменной соли во влажном воздухе. Через несколько дней блестящие поверхности нам будут казаться как бы покрытыми тончайшей пленкой жира. Особенно это будет заметно в сравнении с плоскостями свежих сколов. Наиболее типичными примерами жирного блеска могут служить блеск самородной серы в изломе или блеск элеолита (нефелина), подвергшегося едва заметному разложению.
Поверхности с более грубо выраженной неровностью обладают восковым блеском. Особенно это характерно для скрытокристаллических масс и твердых светлоокрашенных гелей. Таковы, например, часто встречающиеся блески кремней, колломорфных масс минералов группы галлуазита и др.
Наконец, если тонкодисперсные массы вдобавок обладают тонкой пористостью, то в этом случае падающий свет полностью рассеивается в самых различных направлениях. Микроскопические поры являются своего рода «ловушками» для света. Поверхности такого рода носят название матовых. Примерами могут служить: мел, каолин (в сухом состоянии), различные охры, сажистый пиролюзит MnO2, тонкопористые массы гидроокислов железа и т. д.
Для некоторых минералов, обладающих явно выраженной ориентировкой элементов строения в одном или двух измерениях в пространстве, наблюдается своеобразное явление, связанное с блеском, так называемый отлив минерала.
В минералах с параллельноволокнистым строением (асбест, немалит, селенит и др.) мы всегда наблюдаем типичный шелковистый отлив. Прозрачные минералы, обладающие слоистой кристаллической структурой и в связи с этим резко выраженной совершенной спайностью, имеют характерный перламутровый отлив (примеры: мусковит, пластинчатый гипс, тальк и др.). В том, что появление перламутрового отлива связано именно со слоистостью, легко убедиться, если сложить в пачку тонкие покровные или оконные стекла и взглянуть на них сверху. Мы действительно увидим своеобразный отлив, совершенно похожий на блеск жемчужин.
Спайностью называется способность кристаллов и кристаллических зерен раскалываться или расщепляться по определенным кристаллографическим плоскостям, параллельным действительным или возможным граням. Это свойство кристаллических сред связано исключительно с внутренним их строением и для одного и того же минерала не зависит от внешней формы кристаллов (например, у ромбоэдрических, скаленоэдрических и призматических кристаллов или даже совершенно неправильных кристаллических зерен кальцита наблюдается всегда одна и та же форма спайности по ромбоэдру). Этот признак, являющийся характерным для каждого данного кристаллического вещества, служит одним из важных диагностических признаков, помогающих определить минерал. Не случайно многие минералы называются шпатами (полевые шпаты, тяжелый шпат, плавиковый шпат, исландский шпат и т. д.)9. Об этом же говорят названия таких минералов, как ортоклаз (спайность под прямым углом), плагиоклаз (под косым углом) и др.
На практике важно различать степень совершенства проявления спайности. С этой точки зрения принята следующая пятиступенчатая шкала.
1. Спайность весьма совершенная (например, в слюдах и хлоритах). Кристалл способен расщепляться на тонкие листочки (рис. 28). Получить излом иначе, чем по спайности, весьма трудно.
Рис. 28. Весьма совершенная спайность слюды
2. Спайность совершенная (например, в кристаллах кальцита, галенита, каменной соли и др.). При ударе молотком всегда получаются выколки по спайности, внешне очень напоминающие настоящие кристаллы. Например, при разбивании галенита получаются мелкие правильные кубики (рис. 29), при раздроблении кальцита — правильные ромбоэдры и т. п. Получить излом по другим направлениям (не по спайности) довольно трудно.
Рис. 29. Совершенная спайность в трех направлениях и ступенчатый излом галита
3. Спайность средняя (например, в кристаллах полевых шпатов, роговых обманок и др.). На обломках минералов отчетливо наблюдаются как плоскости спайности, так и неровные изломы по случайным направлениям.
4. Спайность несовершенная (например, у апатита, касситерита, самородной серы и др.). Она обнаруживается с трудом, ее приходится искать на обломке минерала. Изломы, как правило, представляют собой неровные поверхности.
5. Спайность весьма несовершенная, т. е. практически отсутствует (например, у корунда, золота, платины, магнетита и др.). Она обнаруживается в исключительных случаях. Такие тела обычно имеют раковистый излом, подобный тому, что наблюдается в изломе шлака или вулканического стекла — обсидиана (рис. 30). Мелкораковистый излом характерен для многих сульфидов. Для некоторых самородных металлов (меди, серебра и др.) характерен занозистый, крючковатый излом.

Рис. 30. Раковистый излом обсидиана
В различных минералах, обладающих спайностью, плоскости последней ориентированы неодинаково для различных типов кристаллических структур: в координационных структурах с ионной связью, например, у галенита (PbS) и галита (NaCl), — по кубу; у кальцита (Са[СО3]) — по ромбоэдру; в силикатах, комплексные анионы которых представлены вытянутыми в одном направлении цепочками, например в пироксенах и роговых обманках, — по призме; в силикатах, характеризующихся анионными слоями, например в слюдах и хлоритах, — по пинакоиду и т. д.
Согласно прежним представлениям, развитым Браве, плоскости спайности проходят параллельно наиболее удаленным друг от друга плоским сеткам пространственной решетки. Г. В. Вульф, основываясь на данных кристаллохимии, показал, что явление спайности в кристаллах с ионной связью обусловлено анизотропией сил сцепления структурных единиц в различных направлениях в кристаллических средах. Так, например, в кристаллической структуре сфалерита (ZnS) наиболее удаленные друг от друга плоские сетки ионов устанавливаются параллельно граням октаэдра, а следовательно, согласно правилу Браве и спайность должна была бы проходить по {111}. На самом деле спайность в сфалерите проявляется параллельно плоскостям ромбического додекаэдра {110}, хотя здесь расстояния между плоскими сетками короче. Дело в том, что в первом случае каждая из удаленных друг от друга плоских сеток сложена одноименными, но разными по заряду ионами (либо Zn2+, либо S2–), что и обусловливает химическую связь между сетками, тогда как во втором случае каждая сетка состоит из взаимно компенсирующих ионов цинка и серы и потому, несмотря на более короткое расстояние, эти плоские сетки слабо связаны между собой. Однако алмаз, обладающий той же кристаллической структурой, что и сфалерит, но состоящий только из атомов углерода, обладает спайностью по октаэдру (т. е. согласно правилу Браве).
Нередко различно ориентированные плоскости спайности в одном и том же минерале имеют различную степень совершенства. Например, у кристаллов гипса, относящихся к моноклинной сингонии, наблюдаются следующие спайности: по второму пинакоиду {010} — весьма совершенная, по ромбической призме {111} — средняя и по первому пинакоиду {100} — несовершенная. Количество направлений спайности в ряде случаев также является важным диагностическим признаком. Например, такие весьма похожие друг на друга по ряду внешних признаков (цвету, твердости, блеску и др.) минералы, как сфалерит — ZnS и вольфрамит — (Fe, Mn)WO4, отличаются друг от друга тем, что в кристаллах или зернах сфалерита наблюдается несколько (шесть) плоскостей спайности по {110}, тогда как у вольфрамита совершенную спайность мы всегда находим только в одной плоскости по {010}, вдоль вытянутости и поперек уплощения кристаллов или зерен.
Кроме спайности, в кристаллах могут наблюдаться также плоскости отдельности, обусловленные, по предположению Н. В. Белова, «прокладками» субмикроскопических веществ иного состава, закономерно ориентированных вдоль плоскостей плотнейшей упаковки. В отличие от спайности они не являются строго плоскими и обычно ориентированы в одном направлении. Причиной проявления отдельности могут быть также внутренние напряжения в кристаллических индивидах, обусловленные внешней механической деформацией или связанные с зональным распределением изоморфных примесей, вызывающим несоразмерность кристаллических решеток в смежных участках кристаллов.
Отдельность, в отличие от спайности, не относится к числу обязательных свойств того или иного минерала, так как не определяется его конституцией. Тем не менее для многих минералов отдельность весьма характерна и проявляется в подавляющем большинстве индивидов, а по качеству образуемых поверхностей может конкурировать с плоскостями спайности (моноклинные пироксены).
Под твердостью10 подразумевают степень сопротивления, которое способен оказать данный минерал какому-либо внешнему механическому воздействию, в частности царапанию.
В обычной минералогической практике применяется наиболее простой способ определения твердости царапанием одного минерала другим, т. е. устанавливается относительная твердость минералов. Для оценки этой твердости принимается шкала Мооса, представленная десятью минералами, из которых каждый последующий своим острым концом царапает все предыдущие.
За эталоны этой шкалы приняты следующие минералы в порядке твердости от 1 до 10:
1) тальк — Mg3[Si4O10][OH]2;
2) гипс — Ca(SO4) . 2H2O;
3) кальцит — Са(СО3);
4) флюорит — CaF2;
5) апатит — Са5[PO4]3F;
6) ортоклаз — K[AlSi3O8];
7) кварц — SiO2;
8) топаз —Al2[SiO4](F,OH)2;
9) корунд — Аl2O3;
10) алмаз — С.
Определение твердости исследуемого минерала производится путем установления, какой из эталонных минералов он царапает последним. Например, если исследуемый минерал царапает апатит, а сам царапается ортоклазом, то это значит, что его твердость заключается между 5 и 6.
Этот простой, хотя и грубый метод определения твердости минералов вполне удовлетворяет нас при диагностике минералов.
В пределах значений по шкале Мооса у большинства минералов на различных гранях и сколах твердость является более или менее постоянной, хотя известны примеры, когда она меняется в зависимости от направления царапания. Например, у минерала кианита Al2[SiO4]O в направлении удлинения твердость равна 4,5, а в перпендикулярном направлении на той же плоскости — 6–7. Поэтому не случайно этот минерал называется также дистеном (от греч. ди — двояко, стенос — сопротивляющийся).
Более точные определения твердости минералов с научно-исследовательской целью производят на специальных приборах — склерометрах — с помощью алмазного или металлического острия. Мы не будем останавливаться на рассмотрении этих устройств. Приведем лишь некоторые выводы, полученные при детальном изучении явлений нарушения поверхностей кристаллов методом царапания.
Прежде всего выяснилось, что твердость кристаллических тел обладает векториальными свойствами (анизотропией), т. е. в различных направлениях в кристалле она не одинакова. Это относится даже к минералам кубической сингонии. В качестве примера на рис. 31 приведена «розетка» твердости на грани куба каменной соли.

Рис. 31. «Розетка» твердости на грани куба каменной соли
Если испытываемая плоскость кристалла ориентирована перпендикулярно плоскости спайности, то в направлении, параллельном следу плоскости спайности, она обнаруживает наименьшие, а в перпендикулярном направлении — наибольшие значения твердости.
С кристаллохимической точки зрения твердость кристаллических тел зависит от типа структуры и прочности связей атомов (ионов). Хотя в этой области мы располагаем еще очень неполными данными, все же некоторые положения установлены с достаточной степенью определенности.
Для ионных кристаллических тел путем сопоставления ряда эмпирических данных выявляется, что твердость, в общем, прямо пропорциональна плотности кристаллических структур. С увеличением межионных расстояний для данного типа соединений она падает:
|
BeO |
MgO |
CaO |
SrO |
BaO |
||
|
Расстояние АХ |
1,55 |
2,10 |
2,40 |
2,57 |
2,77 |
Å |
|
Твердость по Моосу |
9 |
6,5 |
4,5 |
3,5 |
3,5–3 |
Установлено также, что для соединений, кристаллизующихся в одинаковой структуре и с близкими межионными расстояниями, с увеличением валентности, т. е. зарядов ионов, твердость возрастает.
Кроме того, как показал акад. В. С. Соболев, большое значение имеет координация катионов в соединениях. В окислах и силикатах наибольшая твердость принадлежит соединениям тех катионов, для которых отношение rк : rA отвечает нижнему пределу устойчивости координационного числа. Для силикатов, содержащих алюминий в шестерной координации, твердость выше, чем в алюмосиликатах (с четверной координацией Аl). Присутствие в составе соединений гидроксильных ионов и воды несколько снижает твердость.
Следует указать, что скрытокристаллические, тонкопористые и порошковатые разности минералов обладают ложными малыми твердостями. Например, гематит (Fe2O3) в кристаллах имеет твердость 6, а в виде красной охры — меньше 1, что говорит практически об отсутствии сцепления между отдельными частицами в тонкодиспергированной массе гематита.
В целом главная масса природных соединений обладает твердостями в пределах от 2 до 6. Более твердые минералы принадлежат к безводным окислам и силикатам: кварц — SiO2 (твердость 7), касситерит — SnO2 (6–7), корунд— Аl2О3 (9), минералы группы шпинели — MgAl2O4 (8), топаз (8), берилл (7,5–8), турмалины (7–7,5), гранаты (около 7) и др.
Хрупкость, ковкость, упругость
Эти свойства при диагностике минералов имеют второстепенное значение, однако для ряда минералов они весьма характерны.
Под хрупкостью подразумевается свойство минерала крошиться под давлением при проведении острием ножа царапины по его поверхности. При этом, например, минерал, известный под названием «блеклая руда», «пылится», т. е. дает матовую черту с темным порошком по краям. Халькозин, похожий на него по внешним признакам, в этом случае дает гладкий блестящий след, что свидетельствует о его свойстве пластической деформации, т. е. о ковкости. Аналогичное явление, но в более резко выраженной форме обнаруживают ковкие самородные металлы (медь, золото, серебро и др.). Свойство ковкости их проявляется также и в том, что их зернышки на наковальне с помощью молоточка могут быть расплющены в тонкие пластинки
Для ряда минералов, обладающих слабыми кристаллическими структурами, явление пластичности обусловливается скольжением, состоящим в параллельном перемещении слоев кристаллической среды вдоль одной или нескольких плоскостей (скольжения в каменной соли). В других минералах пластичность сопровождается механическим двойникованием (например, в кристаллах кальцита).
Упругость, т. е. свойство вещества изменять свою форму под влиянием деформирующих сил и вновь ее восстанавливать по их удалении, также характерна для некоторых минералов. Этим свойством обладают, например, слюды, чем они отличаются от кальцийсодержащих, так называемых хрупких слюд, ломающихся при изгибе. Похожие на слюды хлориты при сильном изгибе хотя и не ломаются, но не восстанавливают своего прежнего положения. Большинство минералов, способных выделяться в виде асбестов, при их расщеплении дают тончайшее эластичное волокно, поддающееся текстильной обработке. Волокнистая разновидность гипса — селенит — этим свойством не обладает.
Удельный вес (в настоящее время все чаще заменяется термином «плотность», в минералогии традиционно используется единица плотности г/cм3) минералов, как известно, зависит прежде всего от атомного веса атомов или ионов, слагающих кристаллическое вещество. Затем существенную роль играют размеры ионных радиусов, возрастание которых компенсирует увеличение атомного веса, иногда настолько, что удельный вес даже снижается: например, несмотря на то что атомный вес калия в 1,7 раза больше, чем натрия, удельный вес КСl (1,98) меньше, чем NaCl (2,17) в силу того, что ионный радиус К1+ (1,33) больше ионного радиуса Na1+ (0,98), что сильно сказывается на объеме кристаллического вещества. Кроме того, как указывалось выше (см. полиморфизм), изменение, в частности увеличение координационного числа в кристаллических структурах, приводит к уменьшению объема, а следовательно, к увеличению удельного веса. Наконец, уменьшение валентности катиона (или увеличение валентности аниона) при прочих равных условиях, по В. С. Соболеву, должно также сопровождаться увеличением удельного веса.
Удельные веса минералов колеблются в широких пределах: от значений меньше 1 (лед, некоторые органические минералы) до 23,0 (некоторые разности минералов группы осмистого иридия).
Главная масса природных органических соединений, окислов и солей легких металлов, расположенных в верхней части менделеевской таблицы, обладает удельными весами в пределах от 1 до 3,5: янтарь, твердые битумы (1–1,1), галит — NaCl (2,1–2,5), гипс — CaSO4 . 2H2O (2,3), кварц — SiO2 (2,65), алмаз (3,51) и др. Лишь некоторые относящиеся к этой группе минералы имеют больший удельный вес; таковы, например, корунд и его разновидности — Аl2О3 (4). Среди сульфатов особо выделяется барит — BaSO4 (4,3–4,7), почему он и получил свое название (от греч. барос — тяжесть).
Соединения типичных тяжелых металлов, занимающих нижнюю часть менделеевской таблицы, характеризуются средними удельными весами от 3,6 до 9. Примеры: сидерит — FeCO3 (3,9), сфалерит — ZnS (4), пирит — FeS2 (5), магнетит — FeFe2O4 (4,9–5,2), гематит — Fe2O3 (5–5,2), англезит — PbSO4 (6,4), церуссит — РbСО3 (6,5), касситерит — SnO2 (7), галенит — PbS (7,5), киноварь — HgS (8), уранинит — UO2 (8–10).
Наибольшие удельные веса имеют самородные тяжелые металлы: медь (8,9), висмут (9,7), серебро (10–11), золото (15–19), минералы группы самородной платины (14–20), минералы группы иридия и осмистого иридия (17–23).
Удельные веса минералов определяются в основном двумя способами: 1) методом вытеснения жидкости, т. е. путем взвешивания образца и измерения объема вытесненной им воды в сосуде; 2) путем определения потери в весе минерала, погруженного в воду (абсолютный вес образца делят на потерю им веса в воде). Удельный вес мелких зернышек минерала определяется с помощью так называемого пикнометра или тяжелых жидкостей и весов Вестфаля, описываемых в специальных руководствах.
Довольно значительные колебания удельного веса, устанавливаемые для одного и того же минерала, наблюдаются сравнительно редко и, помимо изоморфизма, обычно бывают обусловлены мельчайшими включениями посторонних минералов, в том числе пузырьков газа и жидкостей.
Различие в удельных весах минералов широко используется при обогащении руд различными гравитационными методами с целью отделения нерудных минералов (кварца, кальцита, барита и др.) от рудных минералов (галенита, сфалерита, касситерита и др.). При обогащении получаются концентраты с повышенным содержанием полезных минералов.
Существует очень немного минералов, которые обладают явно выраженными магнитными свойствами. Минералы со слабыми парамагнитными свойствами легко притягиваются магнитом (бедные серой разности пирротина). Но имеются и такие минералы, которые сами представляют собой магнит, т. е. являются ферромагнитными и притягивают к себе железные опилки, булавки, гвозди. Таким свойством обладают магнетит, никелистое железо, некоторые разности ферроплатины. Наконец, известны диамагнитные минералы, отталкивающиеся магнитом (самородный висмут).
Так как число минералов, обладающих магнитными свойствами, невелико, то этот признак имеет важное диагностическое значение. Испытание на магнитность производится c помощью свободно вращающейся магнитной стрелки, к концам которой подносится испытуемый образец. Допускается и употребление магнита, при этом предпочтительно использовать мелкие зерна минерала, а магнит прикрывать бумагой.
Слабыми магнитными свойствами, не устанавливаемыми с помощью магнитной стрелки, обладает довольно большое количество минералов. На различии этих слабо выраженных магнитных свойств основано разделение минералов на фракции с помощью электромагнита при исследовании так называемых шлихов, т. е. тяжелой фракции минералов, получающейся при промывании.
В самом конце периодической системы Менделеева располагается группа радиоактивных элементов урана — радия, представляющих совершенно особый интерес. Явления радиоактивности были открыты А. Беккерелем еще в 1896 г. Дальнейшее изучение их привело к открытию общих законов строения атома и к развитию так называемой ядерной физики.
Как известно, образование ионов, химических соединений и вообще все химические процессы обусловлены почти исключительно строением наружных электронных оболочек атомов и той энергией, которая выделяется при перегруппировках электронов; ядра атомов при этом не претерпевают никаких изменений. Явления же радиоактивных превращений, наоборот, связаны с превращениями, происходящими в самих ядрах. В связи с этим необходимо напомнить о строении атомных ядер.
Установлено, что в строении атома принимают участие три основных вида частиц: протон и нейтрон — в ядре, электрон — в окружении ядра. Число протонов равно атомному номеру, а число нейтронов — разности между массовым числом (близким к атомному весу) и атомным номером. Рассматривая периодическую систему элементов (см. табл. 2), легко видеть, что в первых рядах элементов числа протонов и нейтронов ядра атома обычно равны друг другу. По мере перехода к более тяжелым элементам мы замечаем, что число нейтронов по сравнению с протонами постепенно возрастает (так полагается для достижения минимума энергии, а следовательно, и устойчивости ядра). В последнем ряду устанавливается уже весьма значительный избыток нейтронов по отношению к протонам. Например, ядро тяжелого урана U238 содержит 92 протона и 146 нейтронов (238 – 92), а ядро изотопа U235 — на три нейтрона меньше (при том же числе протонов).
Эти последние элементы периодической системы обладают не вполне устойчивыми ядрами атомов. Для таких элементов весьма характерны явления так называемого радиоактивного распада, выражающиеся в непрерывном испускании:
1) α-частиц, т. е. ядер атомов гелия, обладающих атомным номером 2 и массовым числом 4; они выбрасываются с громадной скоростью и ионизируют воздух, т. е. делают его проводником электричества; испускание этих частиц приводит к тому, что атом данного элемента последовательно превращается в атомы более легких элементов, причем атомный номер при вылете каждой частицы уменьшается на две, а масса — на четыре единицы;
2) β-частиц, равнозначных электронам; испускание одной такой частицы, естественно, приводит к увеличению заряда ядра на единицу (при сохранении массового числа); следовательно, и атомный номер продукта превращения увеличивается на единицу;
3) γ-лучей, представляющих собой электромагнитное излучение, подобное рентгеновским лучам.
Это непрерывное превращение атомов, сопровождающееся большим расходом энергии, протекает вне зависимости от температуры и давления. Конечными продуктами, образующимися в результате последовательных испусканий α- и β-частиц, являются устойчивые изотопы свинца. Скорость распада образующихся промежуточных атомов колеблется в весьма широких пределах от долей секунды до миллиардов лет. Время, необходимое для распада половины всего количества атомов данного изотопа, называется периодом полураспада. Оно постоянно для каждого изотопа.
Установлены три ряда последовательных радиоактивных превращений: 1) ряд урана, начинающийся с изотопа урана U238 (рис. 32), где в числе промежуточных продуктов распада образуется и радий с периодом полураспада 1600 лет; 2) ряд актиния, начинающийся с другого изотопа урана — U235 и включающий в числе промежуточных продуктов превращений актиний; 3) ряд тория, начинающийся с изотопа Th232.
Рис. 32. Начальные участки трех рядов естественных радиоактивных
превращений и искусственно полученные трансурановые элементы —
нептуний и плутоний
Радиоактивность минералов определяется по производимой ими ионизации воздуха с помощью электроскопов, ионизационных камер и различных систем счетчиков. Урансодержащие минералы, способные излучать химически активные лучи, оказывают сильное воздействие также на фотографическую пластинку. Этим пользуются для получения так называемых радиографий. С этой целью отполированный образец руды в темной комнате или в ящике для проявления кладут на фотопластинку на определенное время. Активные лучи в светочувствительном слое производят обычное химическое действие. После проявления места сильного почернения будут указывать на наличие урансодержащих минералов. На позитивном изображении радиографии, т. е. на фотобумаге, светлые участки будут отвечать минералам, богатым радиоактивными элементами, черные — минералам, не содержащим их.
Явление радиоактивного распада, протекающего в течение огромных периодов времени, используется при определении абсолютного геологического возраста различных пород, в которых в свое время образовались радиоактивные минералы. Такое определение возраста возможно прежде всего потому, что скорость распада каждого радиоактивного вещества не только постоянна, но и не зависит ни от температуры, ни от происходящих химических реакций. Вторым важным обстоятельством является то, что содержание конечных продуктов распада (гелия и свинца) минерала находится в прямой зависимости от времени, истекшего с момента образования радиоактивных минералов.
Из многочисленных других свойств минералов (теплопроводность, электропроводность, пироэлектрические и пьезоэлектрические свойства, детекторные свойства, плавкость, растворимость и пр.) укажем лишь те, которые имеют наибольшее диагностическое значение, т. е. в ряде случаев помогают определить минерал или проверить сделанное определение. Упомянем также о тех свойствах, которыми пользуются в своей практике рудокопы.
Такие свойства, как растворимость, плавкость, окрашивание пламени, перлов буры и др., требуют специальной постановки их исследований. Они подробно рассматриваются в курсе о паяльной трубке, и потому здесь мы на них останавливаться не будем. Укажем некоторые другие свойства минералов, которыми иногда пользуются поисковики, горнорабочие и старатели при поисках или разведке месторождений полезных ископаемых.
Запах, издаваемый некоторыми минералами при ударе или разломе, иногда указывает на присутствие тех или иных элементов в руде. Например, самородный мышьяк, арсенопирит (Fe[AsS]) и другие арсениды металлов при резком ударе издают характерный чесночный запах мышьяка, особенно сильно чувствующийся при нагревании и прокаливании на огне. Иногда жильный кварц, с которым бывают связаны минералы редких металлов, при раскалывании выделяет своеобразный неприятный запах, что в рудокопной практике в некоторых случаях является руководящим признаком. У ряда полезных ископаемых различают глинистые запахи и т. д.
Некоторые минералы, особенно в порошковатых массах, могут легко узнаваться на ощупь. Например, всем известный тальк на ощупь кажется жирным, чем отличается от похожего на него пирофиллита. Точно так же порошковатые разности ярозита — KFe3[SO4]2(OH)6 при растирании между пальцами дают ощущение жирного, салящего вещества, что отличает ярозит от охристых, похожих по цвету масс лимонита HFeO2 . aq, кажущихся при той же манипуляции жесткими, песчанистыми.
При определении качества некоторых полезных ископаемых, употребляемых в пищу, прибегают к вкусовым ощущениям, например при поисках и разведке поваренной соли, артезианских питьевых вод и др.
Наконец, некоторую помощь, особенно в рудокопной практике, оказывают звуковые явления. Забойщики в этом отношении нередко обладают виртуозными способностями. Известно, например, что массы церуссита (Рb[СО3]) при падении на пол издают звук, похожий на звук, производимый падением стекла. Точно так же звуки, которые издают в забоях различные по крепости породы и руды при ударах горными инструментами, отличаются друг от друга, что можно заметить лишь при большой практике.
Таким образом, как мы видим, в определении минералов в полевой практике могут принимать участие все пять чувств: зрение, осязание, обоняние, вкус и слух. Исключительную роль играют, конечно, зрение и развивающаяся в результате опыта зрительная память.
1 Брусит, не содержащий железа, в аналогичных условиях вполне устойчив.
2 По В. М. Гольдшмидту, для достижения аморфного состояния в этих случаях не достаточно одной радиоактивности минерала, а необходимы также следующие два условия: 1) первоначально возникающее кристаллическое вещество должно обладать слабой существенно ионной структурой, допускающей перегруппировку или гидролиз; такие решетки образуются преимущественно при соединении слабых оснований со слабыми ангидридами; 2) структура должна содержать один или несколько сортов ионов, способных легко перезаряжаться (например, ионы редких земель) или даже превращаться в нейтральные атомы (например, образование атомарного фтора в флюорите под влиянием радиоактивного излучения со стороны). Сам процесс распада В. М. Гольдшмидт представляет как перегруппировку вещества. Например, соединение YNbO4 превращается в тонкодисперсную смесь (твердый псевдораствор) окислов: Y2O3 и Nb2О5. При такой концепции понятно, почему не наблюдается превращений в аморфное вещество простых соединений, так ThO2 (торианит), или солей сильных кислот со слабыми основаниями, например (Се, La...)PO4 (монацит).
3 За исключением кристаллооптических свойств (светопреломления, двупреломления, плеохроизма и др.), детально излагаемых в курсах кристаллографии и кристалло оптики.
4 Нужно заметить, что в литературе нет единства в понимании этих терминов. Многие авторы термины «облик» и «габитус» считают синонимами.
5 Идиос (греч.) — свой, собственный.
6 Хрома (греч.) — окраска, цвет.
7 Аллос (греч.) — посторонний.
8 Псевдо (греч.) — ложный.
9 К шпатам (от греч. спате — пластина) издавна относят те не имеющие металлического блеска минералы, которые обладают хорошей спайностью в нескольких направлениях.
10 Понятие «твердость тела» до сих пор с определенностью не установлено, несмотря на проводившиеся исследования этого вопроса. Различают твердости царапания, сверления, давления, шлифования. Результаты исследований всех этих методов показывают, что мы, по существу, имеем дело с неодинаковыми по своей природе физическими явлениями.